
 DATOS
DATOS
 BUSCAR
BUSCAR
 ÍNDICE
ÍNDICE
 DESCARGAS
DESCARGAS
 DESARROLLOS
DESARROLLOS
 MODIFICACIONES
MODIFICACIONES
 CONCORDANCIAS
CONCORDANCIAS
 NOTIFICACIONES
NOTIFICACIONES
 ACTOS DE TRÁMITE
ACTOS DE TRÁMITE
 ABOGACÍA
ABOGACÍA
 VIDEOS
VIDEOS
RESOLUCIÓN 62 DE 2006 2007
(marzo 30)
Diario Oficial No. 46.703 de 28 de julio de 2007
INSTITUTO DE HIDROLOGÍA, METEOROLOGÍA Y ESTUDIOS AMBIENTALES
<Resolución derogada por el artículo 35 de la Resolución 63 de 2024>
Por la cual se adoptan los protocolos de muestreo y análisis de laboratorio para la caracterización fisicoquímica de los residuos o desechos peligrosos en el país.
EL DIRECTOR GENERAL DEL INSTITUTO DE HIDROLOGÍA, METEOROLOGÍA Y ESTUDIOS AMBIENTALES, IDEAM,
en uso de sus facultades legales y en especial de las conferidas por la Ley 99 de 1993, Decreto 1277 de 1994, Decreto 1600 de 1994, Decreto 291 de 2004 y Decreto 4741 de 2005,
CONSIDERANDO:
Que de conformidad con lo establecido en la Ley 99 de 1993, artículo 17, el Instituto de Hidrología, Meteorología y Estudios Ambientales, Ideam, es el establecimiento público encargado del levantamiento y manejo de la información científica y técnica sobre los ecosistemas que forman parte del patrimonio ambiental del país, así como de establecer las bases técnicas para clasificar y zonificar el uso del territorio nacional para los fines de planificación y ordenamiento del territorio;
Que corresponde al Instituto efectuar el seguimiento de los recursos biofísicos de la Nación, especialmente en lo referente a su contaminación y degradación, necesarios para la toma de decisiones de las autoridades ambientales;
Que el artículo 5o del Decreto 1277 de 1994, establece que el Ideam es un organismo de apoyo técnico y científico del Ministerio, para lo cual dentro del ámbito de su competencia definirá los estudios, investigaciones, inventarios y actividades de seguimiento y manejo de información que sirvan al Ministerio para fundamentar la toma de decisiones en materia de política ambiental y suministrar las bases para el establecimiento de las normas, disposiciones y regulaciones para el ordenamiento ambiental del territorio, el manejo, uso y aprovechamiento de los recursos naturales renovables;
Que de acuerdo con el Decreto 1600 de 1994, artículo 5o, el Instituto de Hidrología, Meteorología y Estudios Ambientales, Ideam, es la entidad competente para establecer los sistemas de referencia para el sistema de acreditación e intercalibración analítica de los laboratorios cuya actividad esté relacionada con la producción de datos fisicoquímicos y bióticos del medio ambiente en toda la República de Colombia;
Que de conformidad con el parágrafo 2o del artículo 5o, del Decreto 1600 de 1994, los laboratorios que produzcan información cuantitativa, física y biótica para los estudios o análisis ambientales requeridos por las autoridades ambientales competentes, y los demás que produzcan información de carácter oficial relacionada con la calidad del medio ambiente y de los recursos naturales renovables, deberán poseer certificado de acreditación correspondiente otorgado por el Ideam;
Que de acuerdo con el artículo 5o del Decreto 291 de 2004, son facultades del Director General, entre otras, coordinar con el Ministerio de Ambiente, Vivienda y Desarrollo Territorial, las acciones relacionadas con los asuntos institucionales y promover la coordinación de las actividades del Instituto con las demás entidades u organismos públicos que tengan relación con el sector ambiental;
Que conforme al artículo 15 del Decreto 291 de 2004, entre otras funciones le corresponde al Ideam recolectar y generar información sobre uso de recursos naturales renovables, contaminación y degradación por vertimientos, emisiones y residuos sólidos producidos por las diferentes actividades socioeconómicas. Así como desarrollar protocolos, estándares, procedimientos e instrumentos para la captura, almacenamiento, procesamiento y difusión de información sobre el uso de recursos y sobre la generación de vertimientos, emisiones y residuos sólidos producidos por las diferentes actividades socioeconómicas;
Que de acuerdo con el artículo 8o del Decreto 4741 del 30 de diciembre de 2005, por el cual se reglamenta parcialmente la prevención y el manejo de los residuos o desechos peligrosos generados en el marco de la gestión integral:
“Dentro de los doce (12) meses siguientes a partir de la entrada en vigencia del presente decreto, el Ideam definirá los protocolos de muestreo y análisis de laboratorio para la caracterización físico-química de los residuos o desechos peligrosos en el país”;
Que la misma disposición en el parágrafo 2o estipula, que: “Se dará un período de transición de dos (2) años, a partir de la definición de los protocolos de muestreo y análisis por parte del Ideam, para que los laboratorios implementen los métodos de ensayo y obtengan la respectiva acreditación. A partir de ese momento, no se aceptarán resultados de laboratorios que no cuenten con la debida acreditación”;
Que mediante memorando 2007IE3114 del 2 de marzo de 2007, el doctor Carlos Eduardo Gómez Sánchez, Subdirector ( E. ) de Estudios Ambientales, informa que con fundamento en el artículo 8o del Decreto 4741 de 2005, el Instituto realizó la selección de métodos y ensayos apropiados para el país, que permiten la caracterización físico-química de los residuos o desechos peligrosos, que se encuentran recopilados en el documento denominado: “Protocolos para el muestreo y análisis de las características de peligrosidad de los residuos o desechos peligrosos” y que forma parte integral de la presente resolución;
Que por lo expuesto en la parte considerativa, el Director General del Instituto,
RESUELVE:
ARTÍCULO 1o. <Resolución derogada por el artículo 35 de la Resolución 63 de 2024> Adoptar los “protocolos para el muestreo y análisis de las características de peligrosidad de los residuos o desechos peligrosos”, en los siguientes términos:
Indice temático
1. MUESTREO DE RESIDUOS PELIGROSOS
1.1 Diseño para el proceso de muestreo
1.1.1 Tipo de diseño de muestreo
1.1.2 ¿Muestrar sobre tiempo o espacio?
1.2 Tipos de muestras
1.2.1 Muestro parcial vs. muestreo imparcial
1.2.2 Muestras simples vs. muestras compuestas
1.2.3 Muestras de medio vs. muestras de desecho
1.2.4 Muestras homogéneas vs. muestras heterogéneas
1.3. Aproximación estadística para obtención de muestras representativas
1.3.1 Determinación del espaciamiento de la cuadrícula
1.3.2 Determinación del número de muestras a recolectar
1.3.3 Determinación de los puntos de muestreo (método de números al azar)
1.4 Equipos requeridos para dirigirse al sitio de muestreo
1.5 Metodología del muestreo
1.5.1 Selección de las herramientas de muestreo y dispositivos
1.5.1.1 Barriles y costales o bolsas
1.5.1.2 Superficies deprimidas
1.5.1.3 Tanques
1.5.1.4 Tubos, puntos de descarga o puertos de muestreo
1.5.1.5 Depósitos de almacenamiento y cajas rodadoras
1.5.1.6 Pilas de basura
1.5.1.7 Transportadores
1.5.1.8 Estructuras y escombros
1.5.1.9 Suelos superficiales o subterráneos
1.5.2 Selección del dispositivo
1.6 Tipos de muestreo
1.6.1 Muestreo en suelos, sedimentos y otros materiales geológicos
1.6.1.1 Muestras de suelos superficiales recolectados con espátula, pala o cuchara
1.6.1.2 Sólidos o sedimentos recolectados con un taladro manual
1.6.1.3 Sólidos o sedimentos recolectados con un martillo resbalador
1.6.1.4 Sólidos y sedimentos recolectados con un muestreador continuo o una cuchara muestreadora dividida
1.6.1.5 Obtención de muestras sólidas por métodos de presión directa
1.6.1.6 Perforaciones para obtener muestras de roca
1.6.1.7 Recolección de sedimentos por medio de un sistema de dragado
1.6.2 Muestreo en barriles
1.6.2.1 Procedimiento para el muestreo en barriles
1.6.2.2 Preparación de los barriles para el muestreo
1.6.2.3 Apretura del barril para el muestreo
1.6.2.4 Muestreo del barril
1.6.3 Muestreo en tanques
1.6.3.1 Procedimiento para el muestreo en tanques
1.6.3.2 Preparación de los tanques para el muestreo
1.6.3.3 Muestreo del tanque
1.6.3.4 Equipos de muestreo para tanques
1.6.4 Muestreo en pilas de desecho
1.6.4.1 Procedimiento para el muestreo de pilas de desecho
1.6.4.2 Trier
1.6.4.3 Muestreador de arena
1.7 Aseguramiento de calidad y control de calidad
1.8 Cadena de custodia
2. PROTOCOLOS METODOLOGICOS PARA CORROSIVIDAD
2.1 Método de prueba medición electrométrica de pH
2.2 Determinación reserva ácido/álcali
2.3 Método de prueba corrosión al acero
2.4 diagrama metodológico determinación característica de corrosividad
3. PROTOCOLOS METODOLOGICOS PARA EXPLOSIVIDAD
3.1. Prueba para determinar la propagación de la detonación-prueba de excitación con barrera interpuesta
3.2 Prueba de sensibilidad ante condiciones de calor intenso-prueba koenen
3.3 Prueba para determinar el efecto de la inflamación en espacio limitado-prueba de tiempo/presión
3.4 Prueba para determinar el efecto de la inflamación en espacio limitado-prueba de inflamación interior
3.5 Sensibilidad a estímulos mecánicos-choque y fricción
3.6 diagrama metodológico determinación característica de explosividad
4. PROTOCOLOS METODOLOGICOS PARA INFLAMABILIDAD
4.1 Inflamabilidad de líquidos
4.2 Inflamabilidad de sólidos
4.3 Inflamabilidad de gases
4.4 Diagrama metodológico determinación característica de inflamabilidad
5. PROTOCOLOS METODOLOGICOS PARA REACTIVIDAD
5.1 Prueba para sólidos que pueden experimentar combustión espontánea
5.2 Prueba para líquidos que pueden experimentar combustión espontánea
5.3 Prueba para sólidos que pueden experimentar calentamiento espontáneo
5.4 Prueba para sustancias que en contacto con agua desprenden gases inflamables
5.5 Prueba para sustancias sólidas comburentes
5.6 Diagrama metodológico determinación característica de reactividad
6. PROTOCOLOS METODOLOGICOS PARA TOXICIDAD
6.1 Procedimiento de lixiviación para la característica de toxicidad-TCLP
6.2 Procedimiento de lixiviación de precipitación sintética, SPLP
6.3 Toxicidad aguda para Daphnia
6.4 Ensayo de inhibición de algas
6.5 Diagrama metodológico determinación característica de toxicidad
ANEXOS
ANEXO I
DETERMINACION DE MUESTRAS SW-846
ANEXO II
Tabla 1
Guía para la selección de los diseños de muestreo
ANEXO III
Tabla 2
Valores tabulados de “t” para la evaluación de desechos sólidos
ANEXO IV
Tabla 3
Concentraciones estándares-TCLP
ANEXO V
Tabla 4
Guía de selección del dispositivo, medio y sitio de recolección de la muestra
ANEXO VI
Tabla 5
Descripción del medio enumerado en la Tabla 4
ANEXO VII
Tabla 6
Guía de selección del dispositivo-factores específicos del dispositivo
ANEXO VIII
PROCEDIMIENTO PARA LA APLICACION DE CADA DISEÑO DE MUESTREO
ANEXO IX
ESTRATEGIAS PARA EL MUESTREO DE DESECHOS HETEROGENEOS
ANEXO X
DISPOSITIVOS DE MUESTREO
ANEXO XI
PROCESO DE LIXIVIACION PARA MATERIALES DE DESECHO MONOLITICOS “ENSAYO DE TANQUE” (ENSAYO DE DIFUSION)
ANEXO XII
FIGURA EQUIPO EXTRACCION-TCLP/SPLP
ANEXO XIII
FIGURA ZHE-TCLP/SPLP
ANEXO XIV
EQUIPOS CONOCIDOS POR LA EPA, PARA FILTRACION, ZHE-TCLP/SPLP
ANEXO XV
METODOS APROPIADOS PARA ANALISIS DE ANALITOS EN EXTRACTOS
ANEXO XVI
AGUA RECONSTITUIDA
ANEXO XVII
SUSTANCIAS DE REFERENCIA
ANEXO XVIII
RESULTADO DE ENSAYOS INTER-LABORATORIOS CON SUSTANCIA DE REFERENCIA
1. MUESTREO DE RESIDUOS PELIGROSOS
1.1 Diseño para el proceso de muestreo
El muestreo es una de las etapas más críticas de un programa de caracterización física y química de las propiedades de los residuos o desechos peligrosos, por lo tanto requiere de la preparación cuidadosa de plan para la toma y conservación de las muestras.
En esta sección del manual se presenta el desarrollo y la implementación de un plan de muestreo confiable estadísticamente para residuos o desechos peligrosos así como la necesidad de documentar la cadena de custodia para dicho plan. La información presentada en esta sección es importante para el muestreo de residuos o desechos peligrosos que han sido definidos en el Decreto 4741 de 2005, ya sea que se presenten en estado sólido, semisólido, líquido o gaseoso. Sin embargo la gran variedad de estos materiales, así como la gran variedad de facilidades para su disposición (lagunas, pilas, tanques,) y equipos para la toma de muestras exige que cada plan de muestreo sea diferente y muy detallado.
Un plan apropiado de muestreo de residuos peligrosos debe responder a objetivos científicos y objetivos regulatorios. Una vez estos objetivos han sido claramente definidos se debe diseñar un plan apropiado basado en conceptos fundamentales de estadística.
1.1.1 Tipos de diseño de muestreo
En el desarrollo de un proceso de muestreo se consideran varias estrategias para la selección de los sitios, tiempos, y los componentes que se van a muestrear, y se definen apropiadas muestras soporte. Ejemplos de diseños de muestreo incluyen muestreo aleatorio simple, aleatorio estratificado y sistemático.
El diseño de muestreo es el dónde, cuándo y cómo en el proceso de planeación. En el contexto de muestreo de desechos existen dos categorías de diseño de muestreo:
1. Muestreo Probabilística, y
2. Muestreo Autoritativo (no probabilístico).
El muestreo probabilístico se refiere a diseños de muestreo en los cuales todas las partes del desecho o del medio bajo un estudio tienen una probabilidad de estar incluidas en la muestra final. Los muestreos probabilísticos pueden ser de varios tipos, pero de cualquier manera, todos ellos hacen uso de aleatoriedad, la cual permite hacer estamentos estadísticos sobre la calidad de estimativos derivados de datos resultantes.
Existen cinco (5) tipos de diseño del muestreo probabilístico: Muestreo Aleatorio Simple, Muestreo Aleatorio Estratificado, Muestreo Sistemático, Muestreo de Rangos Fijos y Muestreo Secuencial. Una estrategia que puede ser usada para mejorar la precisión de los diseños de muestreo es el procedimiento de muestreo compuesto.
Este tipo de muestreo es una estrategia usada como parte de un diseño de muestreo probabilístico o un diseño de muestreo autoritativo.
1.1.2 ¿Muestrear sobre tiempo o espacio?
Una importante característica de los diseños de muestreo probabilísticas es que ellos pueden ser aplicados a lo largo de una línea de tiempo o de espacio o ambas.
Tiempo
Diseños de muestreo aplicados sobre tiempo pueden ser descritos por un modelo unidimensional que sigue los siguientes pasos:
-- Materiales sólidos sobre una cinta transportadora.
-- Un flujo de líquido, pulpa o lodo moviéndose en un tubo o en un punto de descarga
-- Pilas alargadas continuas.
Espacio
Por razones prácticas, el muestreo de materiales sobre un espacio de tres dimensiones es mejor dirigido a través de materiales que consisten en series de planos de dos dimensiones o grosores más o menos uniformes. Este es el caso para la obtención de muestras de unidades como:
-- Barriles, tanques, superficies comprimidas de desechos con contenidos líquidos de una capa o varias capas
-- Pilas relativamente planas, u otras unidades de almacenamiento
-- Rellenos de tierra, sólidos en una unidad de tratamiento de suelos
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.1. Diseños de muestreo probabilístico sobre espacio o tiempo.
1.2 Tipos de muestras
Los enfoques del muestreo son necesariamente dependientes del tipo de investigación que se va a ser. ¿Es el sitio un vertedero abandonado? ¿Un sitio industrial abandonado? ¿Una laguna u otro cuerpo de agua con contaminantes desconocidos? A continuación se presentan algunos posibles ejemplos de muestras recolectadas:
-- Superficie y suelo subterráneo (usado para caracterizar y clasificar tipos de suelos y ser analizados para determinar la migración vertical u horizontal de contaminantes sospechosos).
-- Desechos grasos.
-- Sedimentos (recolectados desde puntos de deposición sospechosa en ríos, lagos y corrientes).
-- Emanación de gases provenientes de suelos.
-- Agua potable (proveniente de fuentes de suministro de agua para uso residencial y público).
1.2.1 Muestreo parcial vs. muestreo imparcial.
Los métodos de muestreo parcial son usados para recolectar datos sobre sitios particulares en el punto de interés, como áreas de conocida o sospechosa contaminación. Este tipo de muestreo ofrece la determinación de áreas críticas de contaminación. Los métodos de muestreo imparcial (también llamado Muestreo Aleatorio) son usados para recolectar datos que ofrecen una imagen promedio del sitio, aunque este método tiene su mejor aplicabilidad para predecir características generales del sitio. Este método es la selección estadística de los puntos de muestreo.
1.2.2 Muestras simples vs. muestras compuestas.
Muestras simples son muestras sencillas recolectadas desde un sitio específico. Estas muestras ofrecen la mayor información con respecto a la variabilidad espacial del contaminante. Gracias a sus resultados específicos, este método es el más usado en la recolección de muestras en la mayoría de los sitios de investigación. Sin embargo, como cada muestra necesita ser analizada separadamente, los costos de laboratorio para el muestreo simple son más altos que para el muestreo compuesto.
Una muestra compuesta, es catalogada como una mezcla de muestras recolectadas de diferentes sitios o del mismo sitio pero a diferente tiempo. Una muestra compuesta presenta ciertos defectos, los cuales incluyen la dilución de constituyentes químicos de un sitio por muestras de otros sitios. También, estas muestras presentan una dificultad en la identificación del sitio exacto en el cual un contaminante específico reside. Por lo tanto, datos de muestras compuestas ofrecen menos información sobre la variabilidad del contaminante. En el lado positivo, muestras compuestas ofrecen una evaluación cualitativa de las características del área muestreada. También, como las muestras compuestas incluyen la combinación de diferentes muestras en una sola, esto representa un procedimiento menos costoso que el procedimiento de muestreo simple debido a sus reducidos costos en análisis de laboratorio.
1.2.3 Muestras de medio vs. Muestras de desecho.
Una muestra de medio se refiere a un espécimen tomado del ambiente: aire, agua, tierra y biota. Una muestra de desecho se refiere a un espécimen tomado de barriles, tanques, lagunas, hoyos, pilas de desecho o cualquier otra área de disposición de desechos.
Los métodos de muestreo involucran investigación del medio ambiente y áreas específicas de acumulación de desechos. Muestras de desechos son por lo tanto diferentes de las muestras de medio, porque las características generales de los materiales son diferentes, pero también porque los datos requeridos son diferentes.
En la caracterización de los desechos se pregunta:
-- ¿Qué componentes están presentes?
-- ¿Están los contaminantes excediendo cualquier criterio o estándar?
En el caso del medio ambiente también se formulan preguntas:
-- ¿Cuál medio está contaminado?
-- ¿Cuál es el promedio de contaminación?
-- ¿Cuál es la contaminación total (masa, volumen)?
-- ¿Cuál es la contaminación máxima (concentración)?
-- ¿Cuál área del sitio está contaminada?
-- ¿Cuál es la extensión vertical y horizontal del contaminante?
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.2. Muestra compuesta a partir de múltiples muestras.
1.2.4 Muestras homogéneas vs. Muestras heterogéneas.
La composición de las muestras de medio y de las muestras de desechos pueden ser homogéneas o heterogéneas. Una muestra homogénea es aparentemente uniforme en su totalidad, por ejemplo un contenedor almacenando solo arena.
Una muestra heterogénea varía del tipo de contenedor; por ejemplo, una muestra proveniente de un tanque de material, pues esta se separa en una parte aceitosa, acuosa y sólidos suspendidos, o un sólido que exhibe signos de arena, arcilla, roca e incluso escombros de construcción.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.3 Matriz: transportador de contaminantes.
1.3 Aproximación estadística para obtención de muestras representativas
La selección de los puntos de muestreo es realizada estadísticamente. Los métodos estadísticos particulares usados por un proyecto de remediación son seleccionados por la agencia reguladora de cabeza. La agencia puede basar sus decisiones en regulaciones, a menos que existan poderosas razones para escoger otros métodos de muestreo. Un ejemplo de una situación en la cual la agencia puede no escoger métodos estadísticos, puede ser el caso cuando se tiene previo conocimiento de las áreas contaminadas del sitio, en donde se puede hacer un muestreo al azar del sitio no necesariamente costoso. Otro ejemplo podría ser si el sitio presenta barreras para el muestreo al azar como carreteras o edificios, pues en este caso un método de muestreo parcial bien diseñado podría proporcionar la información requerida.
1.3.1 Determinación del espaciamiento de la cuadrícula.
Los sistemas de cuadrícula son usados en el desarrollo de planes de muestreo al azar, en los cuales las muestras están localizadas a distancias constantes de una a otra. Una cuadrícula puede ser rectangular, triangular, o incluso circular si esta es la mejor vía para localizar dónde van a ser recolectadas las muestras.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.4a. Sistema de cuadrícula cuadrada.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.4b. Sistema de cuadrícula triangular.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.4c. Sistema de cuadrícula modificada para explicar la correlación direccional.
El espacio de la cuadrícula o la distancia entre las líneas de la cuadrícula, es seleccionado de acuerdo a los objetivos de muestreo del sitio específico. Esto podría por lo tanto ser planeado para permitir una baja intensidad del muestreo de un sitio preliminar de investigación o para una alta intensidad del muestreo de una investigación correctiva.
Por ejemplo, si se suponen las líneas de la cuadrícula espaciadas en intervalos de 10 pies. Durante el sitio preliminar de investigación, las muestras pueden ser tomadas cada 10 líneas de intersección, resultando una muestra cada 100 pies. Si la revisión de los datos preliminares justifica un muestreo intensivo, este puede entonces ser realizado a lo establecido, es decir cuadrículas de 10 pies. La cuadrícula dibujada en papel o computador, es transferida al sitio actual por los técnicos que miden y estacan el área.
Figura 1.5. Ejemplo de cuadrícula [cada intersección es numerada].
1.3.2 Determinación del número de muestras a recolectar.
El numero de muestras que van a ser tomadas de un contenedor de desechos o sitio puede ser calculado por la aplicación de la fórmula establecida en el documento guía de la EPA SW-846. Esta fórmula, usada en conjunto con el ejercicio modelo planteado en el Apéndice I, puede ser aplicada por las personas sin conocimiento en estadística.
Para aplicar la fórmula, es necesario tener diferentes muestras preliminares analizadas. Los resultados analíticos de esas pequeñas muestras ofrecen los datos necesitados para el uso de la fórmula.
La fórmula para calcular el número de muestras es:
Donde:
n1: | Número mínimo de muestras a tomar. |
t20: | El valor de t es tomado de la Tabla 2 (ver Apéndice III). |
| s2: | Varianza de la muestra. Puede ser calculada siguiendo el procedimiento incluido en el ejercicio modelo. |
RT: | Norma regulativa, concentración determinada por la EPA para ser peligroso, esto es tomado de la Tabla3 (ver Apéndice IV). |
x: Concentración promedio de las diferentes muestras tomadas en el sitio.
1.3.3 Determinación de los puntos de muestreo (método de números al azar).
Una vez se ha determinado el tamaño de las cuadrículas, como también la disposición, y se ha usado la formula previamente sugerida para calcular el número de muestras a recolectar, el siguiente paso es la selección de los objetivos de muestreo para generar estadísticamente un conjunto de números al azar. Estos números, los cuales son dibujados en el papel donde se dibujó anteriormente la cuadrícula que se necesita para la división del sitio, representan los sitios específicos donde los técnicos recolectarán las muestras, los cuales son llamados objetivos de muestreo o puntos de muestreo. El conjunto de números al azar puede, al menos, ser el mismo número de muestras indicadas por la fórmula.
Para generar el conjunto de números necesarios, está disponible una tabla de números al azar (Figura 1.6) o cualquier software diseñado para generar estadísticamente los números al azar necesarios para el muestreo.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.6. Tabla de números al azar.
Uso de la Tabla de números al azar para seleccionar los objetivos de muestreo
Paso 1: Sin mirar, se elige algún punto dentro de la tabla para establecer un punto de partida. El dígito elegido y cualquier dígito contiguo (un número hacia la izquierda) se convierte en el primer número al azar del conjunto.
Paso 2: Continuando con el procedimiento de elección de dígitos establecida en el paso 1, se toman los dos dígitos consecutivos para formar el segundo número al azar; los siguientes dos dígitos más allá serán el tercer conjunto de dígitos. Se continúa hasta que la tabla sea completada.
Paso 3: Si se requieren más números, se puede cambiar la dirección de elección de los números al azar (diagonal o hacia arriba) y se continúa añadiendo números de dos dígitos al conjunto de números. El conjunto de números es completado cuando el número requerido de puntos de muestreo es igual al número obtenido en la fórmula aplicada en el segundo paso (SW-846).
Los números de muestreo seleccionados se grafican en el papel donde inicialmente se dibujó la cuadrícula. En un sitio muy grande las intersecciones de la cuadrícula podrían alcanzar números de tres dígitos, como 001 a 200.
La cuadrícula es entonces transferida al mapa del sitio. Esta cuadrícula es necesaria para determinar dónde señalar las esquinas de la cuadrícula. Usando estas esquinas señaladas, ya se tiene claro la ubicación de los puntos de muestreo.
1.4 Equipos requeridos para dirigirse al sitio de muestreo
Antes de ir al sitio de muestreo, los técnicos deben realizar una lista de equipos. La SW-846 (EPA) presenta una lista de materiales que se deben tener en cuenta durante la preparación del equipo antes de ir al sitio de muestreo. Algunos ejemplos son:
-- Equipo personal: Botas, equipo de lluvia, equipos de protección, máscaras y guantes.
-- Equipo de seguridad: Estaciones de lavado portátiles, equipo de primeros auxilios
-- Equipos de medición en campo: Tubos de muestreo de aire, medidores de pH, indicadores de gas combustible.
-- Equipos de almacenamiento: Recipientes, tapas, y contenedores para acomodar al menos 50% de los requerimientos de muestreo o cambiar el tipo de medio recolectado.
-- Equipos de muestreo: Botella de medición de peso, implementos de descontaminación.
-- Implementos de oficina: Cintas, reglas, formatos, sellos, cuadernos de apunte, tablas de números al azar, tijeras, lápices.
1.5 Metodología de muestreo
1.5.1 Selección de las herramientas de muestreo y dispositivos.
Las herramientas, dispositivos y métodos pueden variar con la forma, consistencia y localización de los materiales de desechos que van a ser muestreados. Como parte del proceso c) Cuando las muestras son recolectadas por espátula, pala o cuchara, use un limpiador para remover residuos de suelo en la herramienta colocando los desperdicios en un recipiente plástico:
d) Empacar las muestras en contenedores. Si un análisis orgánico volátil va a ser ejecutado, transferir una porción de la muestra directamente dentro de un contenedor apropiado con una cuchara metálica o con guantes. Tapar el contenedor hasta el tope, estando seguro que no existan espacios en la parte superior del contenedor. Inmediatamente marque la muestra. Para otros tipos de muestras simples sitúe una porción de suelo dentro de una vasija metálica limpia o balde y mezcle hasta obtener una muestra homogénea. Si después de la mezcla la muestra parece ser homogénea, transferir la muestra a un contenedor. Ubique la muestra dentro de un contenedor apropiado con los preservativos químicos apropiados. Si la muestra es compuesta, ubicar las muestras desde todos los puntos de muestreo o intervalos dentro de un contenedor y mezclar hasta homogenizar el material. Después de que la mezcla es completada, colocar la muestra en un contenedor con tapa. Marca el contenedor;
e) Completar los formatos con los datos de las muestras recolectadas;
f) Colocar todas las muestras dentro de un cuarto frío o algún dispositivo similar a 4 ºC;
g) Rellenar los hoyos abiertos con el material almacenado;
h) Limpiar y restaurar el sitio de muestreo;
i) Descontaminar los equipos y el sitio de muestreo;
j) Antes de abandonar el sitio, anotar los puntos donde se tomaron las muestras y revisar todo lo anotado.
1.6.1.2 Sólidos o sedimentos recolectados con un taladro manual.
Un taladro manual es una columna de acero inoxidable con una terminación en forma de T en la parte superior conectado con sistema giratorio, con un aparato cortador situado al final de la herramienta. Diferentes tipos de taladros manuales están disponibles dependiendo del tipo de material que va a ser muestreado y del tipo de muestra que se requiere.
Un taladro puede ser de tipo cúbico, el cual tiene un recipiente cilíndrico en la base que recoge y retiene el sólido. Algunos taladros cilíndricos como un taladro de barro o un taladro de arena, pueden ser diseñados para trabajar mejor con ciertos tipos de sólidos y sedimentos. Los taladros de aire continuo son de forma espiral y parecen como un sacacorchos. Este taladro puede cavar un hoyo hacia abajo a cierto nivel en donde un tubo de muestreo delgado puede ser introducido hacia el fondo del suelo para recolectar la muestra.
El muestreo con taladros manuales es usualmente limitado a profundidades de 20 pies o menos. La profundidad a la cual el taladro puede efectivamente ser usado depende en el tipo de material como también las capacidades de las personas que están manejando el dispositivo. El hallazgo de aguas superficiales o la presencia de agua en los suelos, pueden causar colapsos en los hoyos. Las gravas son difíciles para ser muestreadas con un taladro manual, pero no es imposible. Otros materiales como escombros, pueden también evitar que el taladro llegue a la profundidad deseada. En general, los taladros manuales son dispositivos de muestreo efectivos solo donde el suelo es fácilmente excavado y el hoyo resultante no colapsa ni se destruye.
En el procedimiento para su aplicación, la parte inicial es similar a lo explicado anteriormente, pero varia inmediatamente después de que se corta el césped, ya que seguido a este paso se introduce el taladro aplicando un presión hacia abajo y rotándolo. Así se retiene la muestra en la parte inferior del dispositivo y después es transferida a un contenedor adecuado.
1.6.1.3 Sólidos o sedimentos recolectados con un martillo resbalador.
Un martillo resbalador es otra herramienta manual para la recolección de muestras a poca profundidad. La herramienta es un martillo resbaladizo localizado en la parte superior del dispositivo, el cual es conectado en el centro de un tubo de acero inoxidable para recoger la muestra. El martillo resbaladizo es usado para manejar el centro del tubo de acero dentro del suelo. Extensiones pueden ser adicionadas para recolectar muestras más profundas.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.7. Diferentes taladros usados para diferentes tipos de suelos.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.8. Martillo resbalador.
1.6.1.4 Sólidos y sedimentos recolectados con un muestreador continuo o una cuchara muestreadora dividida.
Cucharas divididas y muestreadores continuos son dispositivos de muestreo manejados en el suelo dentro de una perforación hecha por una torre perforadora. Estos dispositivos penetran a grandes profundidades.
La cuchara muestreadora dividida es un tubo de acero inoxidable que se abre en dos mitades a lo largo del eje del dispositivo. La cuchara está sujeta al final de una varilla la cual está en el taladro y es bajada hasta el fondo de la perforación. La perforación ha sido previamente taladrada para la profundidad deseada usando un taladro hueco dentro del cual la cuchara es introducida para ser hundida en el suelo. Una vez, en el fondo de la perforación, la cuchara es martillada dentro del suelo usando un peso (comúnmente 140 libras) dentro del taladro. Los técnicos deben anotar:
1. El número de golpes (un peso lanzado desde una altura de 30 pulgadas) necesarios para introducir la cuchara dentro del suelo, y
2. El número de golpes requeridos para mover la cuchara cada 6 pulgadas.
El muestreador continuo es otro tubo de acero inoxidable que se abre en dos mitades longitudinalmente. Este puede tener varios diámetros (diámetros de 2 y 3 pulgadas son comunes) y también varían en longitud. El muestreador continuo es situado al final de la barra en la torre de perforación y es bajado al fondo de la perforación por el taladro hueco. La torre de perforación empuja el muestreador dentro del suelo.
Ambos dispositivos son recuperados desde la perforación y abiertos longitudinalmente, colocando la muestra dentro de cada dispositivo.
1.6.1.5 Obtención de muestras sólidas por métodos de presión directa.
Los métodos de presión directa usan técnicas en donde ciertas herramientas se empujan dentro del suelo sin el uso de taladros. Un tipo de sistema de presión directa es conocido como Geoprobe®. Esta es una máquina hidráulicamente manejada que combina el peso del vehiculo. El Geoprobe® es sujetado a un equipo de percusión por encima de 2000 golpes por minuto para manejar los equipos de muestreo dentro del suelo. El Geoprobe® es típicamente usado para investigaciones a profundidades de 60 pies. Se han encontrado casos donde se ha utilizado en profundidades de más de 100 pies. También recolecta muestras sólidas a intervalos discretos usando una varilla muestreadora larga que tiene 2 pies de longitud y 1.375 pulgadas de diámetro.
1.6.1.6 Perforaciones para obtener muestras de roca.
Una perforación es requerida para obtener muestras o corazones de rocas profundas. Diferentes métodos de perforación están disponibles, fragmentos y/o corazones de rocas pueden ser obtenidas por este método. A continuación se presenta una breve descripción de algunos métodos de perforación.
Perforación rotatoria. Una broca en el final del taladro es girada en contra del material de la roca para formar el hoyo. Como la broca y el tubo son rotados, el fluido de circulación es bombeado dentro del hoyo para limpiar los cortes a la superficie y para refrescar y lubricar la broca Perforación de percusión. La broca es martillada o golpeada en la roca para forzar la roca al rompimiento.
Perforación por cable. Es un tipo de perforación de percusión; es lenta pero tiene una ventaja de no requerir fluido para sacar el material fuera del hoyo. Con una perforación de percusión al aire, la energía del aire, presiona el martillo hacia abajo y transporta los cortes a la superficie del hoyo. Un método similar de perforación de percusión al aire, es el martillo operado por aire, el cual usa una segunda herramienta para arrastrar los cortes de roca a la superficie. Ambos métodos de perforación de percusión al aire requieren una lubricación con petróleo en línea de las partes internas.
Muestras recolectadas como fragmentos son colocadas dentro de contenedores-bolsas o botellas-y marcadas de acuerdo al punto de recolección, profundidad e información relacionada con la muestra.
Muestras recolectadas como corazones o centros de rocas son registradas y localizadas dentro de cajas especiales o tubos.
1.6.1.7 Recolección de sedimentos por medio de un sistema de dragado.
Algunas veces una muestra es requerida desde un área sumergida. El agua puede ser poco profunda (solo unas pocas pulgadas) o muchos pies de profundidad. Esto puede ser un cuerpo de agua estática como un estanque o un lago, o una corriente de agua como un río o un riachuelo. Los sedimentos que están por debajo del agua pueden ser recolectados directamente con el uso de un dispositivo manual como una pala o taladro, o indirectamente usando un sistema de dragado. El método usado para la recolección de las muestras depende en la profundidad del agua a la que se encuentra el sitio de muestreo y las características físicas de sedimento que va a ser muestreado.
1.6.2 Muestreo en barriles.
1.6.2.1 Procedimiento para el muestreo en barriles.
-- A causa de que los barriles son considerados potencialmente peligrosos, es indispensable conocer los planes de salud y seguridad en el sitio.
-- Antes de comenzar a manejar un barril, es importante inspeccionar cualquier aviso o marcas.
-- Aunque los niveles indicados expresan que no es peligroso, los técnicos deben ser cautelosos, ya que una clasificación del barril algunas veces no especifica de manera correcta sus características.
-- La inspección puede incluir un monitoreo alrededor de los barriles con un medidor de radiación, un indicador de gas combustible y un monitor de toxicidad.
1.6.2.2 Preparación de los barriles para el muestreo.
Los barriles pueden ser preparados para un fácil acceso, lo que significa colocarlos de una manera ordenada y de manera vertical permitiendo un espacio para que los técnicos puedan tomar las muestras. En el sitio de preparación, los barriles son separados físicamente en categorías de acuerdo a sus contenidos; barriles con contenido líquido, barriles con contenedores en su interior, como botellones (contenedores plásticos o de vidrio diseñados para mantener materiales cáusticos que pueden reaccionar con metales) y paquetes de laboratorio (barriles usados para mantener pequeños contenedores con químicos que pueden reaccionar peligrosamente con otros); o cilindros de gas.
Barriles sospechosos con materiales explosivos o inflamables deben ser constantemente monitoreados con un medidor de oxígeno/CGI durante su preparación.
1.6.2.3 Apertura del barril para el muestreo.
El área apertura del barril puede ser físicamente separada de las áreas de eliminación y preparación del barril. La apertura de los barriles puede ser con:
1. Con una llave inglesa manual, o
2. Con un dispositivo remoto que perfora o remueve la tapa.
Apertura manual de los barriles.
-- Llave inglesa manual de tapón. Una llave inglesa universal tiene accesorios para remover tapones. Para manejar este, los técnicos colocan el barril verticalmente con el tapón hacia arriba o, para barriles con tapones a los lados, acostar el barril con el tapón hacia arriba. Una barra larga puede ser adherida para mejorar el manejo de la llave.
-- Descabezado manual. Este dispositivo funciona abriendo la tapa como si se estuviera cortando. Es ubicado con sus cuchillas justo en la parte superior o en el borde del barril, luego se hace una presión en contra del lado del barril. Moviendo la manivela de arriba hacia abajo se puede cortar totalmente la tapa y así el barril queda abierto.
Apertura Remota de los barriles.
Destapadores remotos, los cuales son más costosos pero también más seguros para los técnicos, incluyen una punta en forma de garabato, el destapador hidráulico y el destapador neumático.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.9. Dispositivo de apertura remota.
-- Punta en forma de garabato.
Una punta de metal atada a un elemento móvil es bajada para perforar la parte superior del barril. La punta debe ser descontaminada antes de abrir cada barril.
-- Destapador hidráulico de barril.
El destapador hidráulico tiene una bomba la cual presuriza aceite a través de una línea hidráulica de longitud. Un dispositivo metálico penetrante es atado al final de la bomba hidráulica penetrando el barril. El dispositivo de penetración puede ser ubicado, ya sea en la parte superior del barril o a un lado de este.
-- Destapador de tapón neumático.
Un tapón neumático elimina aire comprimido repartiéndolo a través de una línea de aire a un taladro neumático adaptado a un tapón apropiado. El tapón es mantenido por un sistema de agarre adecuado. Una vez el tapón ha sido aflojado, el sistema de agarre es removido antes de realizar el muestreo del barril.
1.6.2.4 Muestreo del barril.
Cada barril seleccionado para el muestreo debe ser monitoreado antes y durante de la actividad. A continuación se presentan 2 de los dispositivos más usados para el muestreo de barriles.
Vaso o tubo hueco.
Es un tubo desechable de 6 a 16 mm de diámetro y 1.2 metros de longitud. El procedimiento para su uso es el siguiente:
-- Remover el material de protección del contenedor que va a ser muestreado.
-- Insertar el tubo hasta el fondo del barril o hasta que una capa de sólidos sea encontrada.
-- Permitir que el desecho dentro del barril alcance su nivel natural dentro del tubo.
-- Tapar la parte superior del tubo con un tapón o el dedo pulgar, estando seguro que el líquido no entre en contacto con el tapón.
-- Cuidadosamente remover el tapón del tubo desde el tanque, insertar el extremo destapado en un envase de muestra.
-- Soltar el tapón y permitir que el tubo se descargue. Descargue cualquier material remanente dentro del barril.
-- Remover el tubo del contenedor muestreado, partirlo en dos piezas, y colocar las piezas en el barril de disposición.
-- Resellar el barril y descontaminar el equipo.
Muestreador de desechos líquidos compuestos-Coliwasa.
Este dispositivo es un vaso o tubo de polipropileno con un tapón en su punto más bajo, el cual permite tomar muestras representativas de desechos heterogéneos en tanques y barriles. El tapón es atado a una barra que se desliza hacia arriba a través del tubo. Algunos Coliwasa son destinados para ser desechables. Su fabricación los hace adecuados para muestrear líquidos. El procedimiento para el uso del Coliwasa es el siguiente:
-- Con el tapón en posición abierta, lentamente bajar el Coliwasa dentro del barril o tanque permitiendo que el nivel del agua sea el mismo adentro y afuera del tubo. Si el nivel del líquido dentro del Coliwasa es más bajo que el nivel de afuera, la toma de muestra va a ser muy rápida y resultará una muestra no representativa.
-- Cuando el tapón choca con el fondo del contenedor del desecho, presionar el Coliwasa hacia abajo contra en tapón para cerrarlo. Cierre el tapón.
-- Lentamente retirar el Coliwasa del contenedor del desecho con una mano mientras que con la otra mano limpiar el tubo con un trapo desechable.
-- Cuidadosamente descargar la muestra dentro de un contenedor de desechos presionando el tapón.
-- Resellar el tanque y descontaminar el equipo.
1.6.3 Muestreo en tanques.
1.6.3.1 Procedimiento para el muestreo en tanques.
Un tanque puede ser un buque carguero (o un camión o tren), una pila conteniendo químicos provenientes de procesos de manufacturación o estructuras para el almacenamiento de químicos o desechos. Los tanques pueden contener materiales peligrosos o no peligrosos o materiales de desecho en forma de líquidos, lodos o sólidos de varias estructuras.
1.6.3.2 Preparación de los tanques para el muestreo.
La EPA enumera los siguientes pasos para preparar los tanques para el muestreo:
-- Inspeccionar las características exteriores del tanque y anotar las observaciones. Los puntos de muestreo potenciales deben ser evaluados para seguridad, accesibilidad y calidad de la muestra.
-- Antes de la inspección interna del tanque, el equipo que realiza el muestreo debería:
1. Revisar procedimientos de seguridad y planes de contingencia de emergencia.
2. Asegurar que el tanque está debidamente situado en el suelo.
3. Remover todas las fuentes de ignición del área inmediata.
-- Utilizar herramientas para remover cualquier cobertura.
-- Recolectar medidas de la calidad del aire en cada sitio de muestreo. Tomar lecturas desde la parte superior del tanque, desde encima del puerto de muestreo y en la zona de respiración.
-- Antes de iniciar el proceso de muestreo, usar un ventilador para limpiar cualquier concentración de tóxico o explosivo presente en la parte superior del barril. No iniciar el proceso del muestreo hasta que las lecturas de los límites de explosión sean más bajos que el 10%.
1.6.3.3 Muestreo del tanque.
Una vez se ha determinado que el tanque es seguro para el muestreo, los técnicos deben seguir los siguientes pasos:
-- Determinar la profundidad del líquido, lodo o sólido
-- Desde la parte superior del tanque, usar un muestreador para muestras subterráneas o una bomba muestreadora para recolectar muestras líquidas. Para muestras representativas líquidas de más de 5 pies de profundidad, recolectar desde 1 pie debajo de la superficie, desde la profundidad media del líquido, y desde 1 pie encima del lodo. Para líquidos a menos de 5 pies, recolectar una muestra representativa con el Coliwasa o tubo hueco. Si el tanque tiene más de un componente, recolectar muestras directamente de cada capa.
-- Visualmente comparar las tres muestras líquidas (o la muestra del Coliwasa) para determinar si estas indican diferentes estratos o fases. Si diferentes fases aparecen en las tres muestras separadas, muestras adicionales deben ser tomadas entre cada punto de muestreo para determinar dónde ocurren los cambios entre cada estrato.
-- Si otro punto de muestreo está disponible, repetir el procedimiento de muestreo para verificar las fases.
-- Volver a colocar la cobertura al tanque.
-- Medir el diámetro exterior del tanque y calcular el volumen de desecho usando las medidas de profundidad.
-- Anotar toda la información. Marcar el contenedor.
-- Descontaminar el equipo de muestreo.
1.6.3.4 Equipos de muestreo para tanques.
Otros equipos usados para el muestreo de tanques son:
-- Cinta métrica.
-- Peso.
-- Cámara filmadora.
-- Baldes.
-- Linterna.
-- Indicador del nivel de agua y aceite.
-- Indicador de gas combustible.
Bomba muestreadora.
Este dispositivo puede recolectar una muestra desde niveles específicos en un tanque de almacenamiento. Este es un cilindro de acero inoxidable hueco con un peso hundido que actúa como una válvula que permite el muestreo. Una cuerda atada en la parte superior del peso abre y cierra la válvula, una segunda cuerda es atada para remover la cobertura la cual ha sido cerrada mecánicamente para mantener la válvula cerrada después del muestreo. El procedimiento para el uso de la bomba muestreadora es el siguiente:
-- Atar la cuerda para la toma de la muestra y la cuerda para cerrar la válvula a la bomba muestreadora.
-- Medir y marcar la cuerda para la toma de la muestra a la altura deseada.
-- Gradualmente bajar la bomba muestreadora con la cuerda hasta alcanzar la altura deseada.
-- Cuando el nivel deseado es alcanzado, halar la cuerda de la válvula para permitir que el muestreador se llene, y luego soltar la cuerda para resellar el muestreador.
-- Retirar la bomba muestreadora con la cuerda.
-- Limpiar la bomba.
-- Ubicar el muestreador sobre el contenedor de la muestra y resellar su contenido.
Muestreador de Lodo.
Este dispositivo puede recolectar muestras de un tanque grande (15 pies de profundidad). Está fabricado con tubo plástico de ¾ de pulgada dividido en secciones de 5 pies marcadas cada pie. Las secciones tienen roscas que permiten que el lodo sea extendido. La parte superior tiene un nylon para alcanzar el muestreador. La parte inferior tiene una válvula de control con un balón que flota hacia arriba mientras que el sedimento es hundido y hacia abajo mientras que el sedimento es alcanzado. El procedimiento para su adecuado uso es:
-- Bajar el muestreador de lodo hasta el fondo del tanque.
-- Cuando la profundidad ha sido alcanzada y el tubo ha sido llenado hasta el nivel de la superficie, arrastrar ligeramente la cuerda como si se estuviera recuperando el dispositivo
-- Cuando la unidad ha sido colocada en el líquido del tanque, la cantidad de lodo en la muestra puede ser leída usando las divisiones marcadas en el tubo.
Muestreador de materiales simples subterráneos.
Este dispositivo es un tubo construido de acero inoxidable o aluminio con una longitud de 6 pies con un montaje de polipropileno o Teflon® en su parte inferior. Un cable que va desde la parte superior del dispositivo hasta la inferior permite abrir y cerrar el contenedor a profundidades específicas. Algunos modelos ofrecen tubos de 6 pies de extensión que permiten tomar muestras de hasta 30 pies o más de longitud. El procedimiento para el uso de este dispositivo es el siguiente:
-- Atornillar la botella muestreadora en la parte superior del dispositivo.
-- Bajar el muestreador a la altura deseada.
-- Colocar el aro en la parte superior del tubo, el cual abre el tapón cargado en la cabeza del montaje.
-- Cuando la botella está llena, liberar el anillo, levantar el muestreador y remover la botella muestreadora.
1.6.4 Muestreo en pilas de desecho.
1.6.4.1 Procedimiento para el muestreo de pilas de desecho.
Las Pilas de desecho con contenidos desconocidos o heterogéneos son comunes en sitios de desechos peligrosos incontrolados. Para obtener una muestra representativa de una pila de desecho heterogéneo, el plan de muestreo debería especificar el muestreo aleatorio estratificado aproximado. El muestreo aleatorio estratificado, el cual está basado en una cuadrícula tridimensional y métodos de números al azar, requiere que los técnicos tengan acceso dentro de la pila de desecho. Una representación adecuada de una pila puede ser reunida desde diferentes muestras al azar. Si el muestreo es limitado en ciertos puntos de la pila, un muestreo basado estadísticamente puede ser representativo solo en la porción muestreada, a menos que el desecho sea homogéneo. El muestreo de pilas de desecho es similar al muestreo de sólidos, por ejemplo, palas, cucharas, taladros manuales, tubos muestreadores. Triers y muestreadores de arena pueden ser usados para sustancias granulares.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 1.10. Cuadrícula de una pila de desecho o un contenedor. Figura 1.10. Cuadrícula de una pila de desecho o un contenedor.
1.6.4.2 Trier.
Este dispositivo está formado de un tubo de acero inoxidable que se corta a lo largo de un lado. Este tubo tiene una terminación en forma de T en un lado y es puntiagudo en el otro lado. Es introducido dentro de la pila de desecho y es girado como un martillo para extraer una muestra representativa. El procedimiento para el uso del Trier es el siguiente:
-- Insertar el Trier dentro del material que va a ser muestreado con un ángulo de 0 o a 45º.
-- Rotar uno o dos veces el Trier para cortar un buena porción de muestra.
-- Lentamente retirar el Trier.
-- Para análisis de orgánicos volátiles, transferir la muestra a un contenedor apropiado con una cuchara y asegurar con una tapa. Colocar el remanente de muestra en un contenedor homogenizador para mezclar completamente la muestra. Situar la mezcla en un contenedor y taparlo, o si se tienen muestras compuestas, situar otra muestra de otro sitio de muestreo en el contenedor homogenizador y mezclar ambas muestras. Cuando la mezcla es terminada, situar las mezclas en un contenedor.
1.6.4.3 Muestreador de arena.
Consiste de dos latas con ranuras o tubos de acero inoxidable. El exterior del tubo tiene una punta puntiaguda para penetrar el material que está siendo muestreado. El procedimiento para el uso adecuado de este dispositivo es el siguiente:
-- El muestreador en posición cerrada se debe insertar dentro del material granular o polvo o el desecho que está siendo muestreado desde un punto cerca del borde o de la esquina, a través del centro, y a un punto diagonalmente opuesto al punto de entrada.
-- Rotar el tubo en posición abierta.
-- Menear el tubo para que el material entre por las ranuras abiertas.
-- Cerrar el muestreador y retirarlo del material.
-- Colocar el muestreador en posición horizontal con las ranuras hacia arriba.
-- Rotar el tubo exterior y deslizarlo desde el tubo interior.
-- Transferir el material a un adecuado contenedor.
1.7 Aseguramiento de calidad y control de calidad
El aseguramiento de calidad se define como el proceso para asegurar que todos los resultados y decisiones basadas en estos resultados son técnicamente confiables y estadísticamente válidos y son documentados apropiadamente. Los procedimientos de control de calidad son herramientas empleadas para medir el grado de cumplimiento de los objetivos de aseguramiento de calidad.
Los resultados no se pueden evaluar con respecto a la precisión y exactitud si no están acompañados de los datos de aseguramiento de la calidad. Estos datos resultan de la implementación de procedimientos de control de calidad durante el muestreo y análisis.
Las principales estrategias para documentar la precisión y la exactitud son:
1.7.1 Blanco de viaje. Se utilizan para detectar algún tipo de contaminación durante el manejo y transporte de la muestra. Este blanco de viaje debe acompañar a las muestras hacia y desde el campo.
1.7.2 Blanco de campo. Son alícuotas libres de metales y compuestos orgánicos que en contacto con los equipos de muestreo son analizadas para detectar algún tipo de contaminación procedente de los equipos de muestreo o contaminación cruzada originada previamente a la recolección de la muestra y durante la toma de la muestra.
1.7.3 Duplicados de campo. Se emplean para documentar la precisión. El valor de la precisión es función de la variación en la composición de la muestra, la variación en la técnica de muestreo y variación de la técnica analítica.
1.7.4 Adicionados de campo. Son usados para determinar la pérdida del analito de interés durante el muestreo y transporte hasta el laboratorio. Dado que el adicionado de campo a veces se prepara en el campo, esto puede conducir en errores por pérdida o por contaminación. Para eliminar este problema es aconsejable preparar en el laboratorio estos adicionados a blancos o a muestras similares a los residuos y luego transportarlos a lo largo del muestreo en los mismos recipientes que contienen las muestras.
1.7.5 Adicionalmente al control de calidad explicado anteriormente, el programa completo de aseguramiento de calidad debe existir el protocolo para la toma de muestras que considere todos los aspectos esenciales de este proceso y debe contener los siguientes aspectos:
a) Definición del objetivo de muestreo;
b) Definición del plan de muestreo;
c) Preparación de los recipientes y equipos;
d) Mantenimiento, calibración, limpieza de los equipos de campo;
e) Preservación, empaque y transporte de muestras;
f) Procedimientos de seguridad industrial y de salud ocupacional;
g) Procedimientos de cadena de custodia.
El labaoratorio debe mantener los siguientes registros:
Nombre y título de los analistas que ejecutan los análisis y el encargado de control de calidad que verifica los resultados y los cuadernos manuscritos del analista y del equipo en los que se contengan los siguientes datos:
a) Identificación de la muestra;
b) Fecha de análisis;
c) Cantidad de muestra utilizada;
d) Número de muestras de control de calidad analizadas;
e) Trazabilidad de las calibraciones de los instrumentos de medición;
f) Evidencia de la aceptación o rechazo de los resultados;
g) La información original reportada por los equipos en disquetes o en otros respaldos de información.
1.8 Cadena de custodia
Una parte esencial de cualquier esquema de muestreo y análisis es asegurar la integridad de las muestras desde la toma hasta el reporte de los resultados. La posesión y el manejo de las muestras deben ser trazables desde el tiempo de toma de la muestra, etapas de análisis y descarte de la muestra. Esta documentación de la historia de la muestra se denomina cadena de custodia. Esta documentación es importante y necesaria cuando existe la posibilidad de que los resultados analíticos o las decisiones basadas en estos resultados podrán ser usados en litigios jurídicos.
La cadena de custodia debe considerar la identificación de la muestra por medio de rótulo que debe contener:
a) Número de la muestra;
b) Nombre de la persona que toma la muestra;
c) Fecha de toma de muestra;
d) Lugar de toma de la muestra.
Toda la información pertinente al trabajo de campo se debe almacenar en un libro foliado consecutivamente y debe contener la siguiente información:
a) Localización del sitio de muestreo;
b) Nombre y dirección de la persona que toma la muestra;
c) Nombre de la empresa que produce el residuo, si es diferente del sitio de muestreo;
d) Tipo de proceso que produce el residuo;
e) Tipo de residuo;
f) Composición aproximada del residuo peligroso;
g) Número y volumen de la muestra tomada;
h) Propósito del muestreo (Vigilancia, contrato);
i) Descripción del sitio de muestreo y del método de muestreo;
j) Fecha y hora de la toma de muestra;
k) Identificación de la persona que toma la muestra;
l) Transporte y distribución de la muestra (nombre del laboratorio, UPS, Federal express);
m) Referencias tales como mapas, fotografías del sitio de muestreo;
n) Observaciones de campo;
o) Mediciones hechas en campo (pH, inflamabilidad, explosividad);
p) Firma del personal responsable de las observaciones.
La hoja de solicitud de análisis es necesaria para acompañar la entrega de la muestra al laboratorio. La porción de campo de este formato debe ser llenada por la persona que tomó la muestra y debe contener la información pertinente anotada en el libro de campo. La porción del laboratorio de este formato debe ser completado por el personal del laboratorio y debe incluir:
a) Nombre de la persona que recibe la muestra;
b) Número de la muestra en el laboratorio;
c) Fecha y hora de recibo de la muestra;
d) Entrega de la muestra;
e) Análisis requeridos.
La muestra debe ser entregada al laboratorio para análisis en el menor tiempo posible; no más de 2 días después del muestreo, la muestra deberá estar acompañada de la cadena de custodia y el formato de solicitud de análisis. La muestra debe ser entregada en el laboratorio a la persona autorizada para recibir muestras.
Las muestras de residuos peligrosos deben ser transportadas de la siguiente manera:
a) Recoger la muestra en recipientes de 16 onzas o más pequeñas de vidrio o polietileno con tapa de teflón, para líquidos dejar espacio suficiente, aproximadamente el 10%. Si el material es sólido, el recipiente más el contenido no deben exceder de una libra de peso. Si se está tomando una muestra de compuestos orgánicos volátiles, llene el recipiente hasta el septun, pero coloque este recipiente dentro de uno de 16 onzas o menos de tal forma que se suministre el espacio de aire requerido. Para cantidades mayores por encima de un galón, se puede recolectar si el punto de inflamación de la muestra es de 23 ºC o más. En este caso el punto de inflamación debe ser marcado en la parte exterior del recipiente y el papel de empaque debe indicar que el punto de inflamación es de 23 ºC o más.
b) Sellar la muestra y colocarla en una bolsa de polietileno 4-mil-tick, una muestra por bolsa;
c) Coloque la bolsa sellada dentro de una caja metálica con material absorbente no combustible (Vermiculita o tierra), para evitar que se rompa, una bolsa por caja. Cierre a presión la caja, use clips y asegure la tapa;
d) Marque la caja con:
Nombre y dirección del generador del residuo.
Colocar la nota “Líquido inflamable, NOS UN 1993”.
o (sólido inflamable, NOS UN 1325);
e) Coloque una o más cajas de metal en un contenedor robusto como neveras de camping. No se usan sustancias preservantes para muestras de residuos peligrosos en campo;
f) Preparación para el embarque: las palabras nota “Líquido inflamable, NOS UN 1993” o (sólido inflamable, NOS UN 1325, Cargo Aircraft Only; Limited Quantity, Laboratory
Simples, se deben indicar en los papeles de embarque en la parte exterior del contenedor. También se deben colocar las palabras This Side UP o This Side End UP. Firmar la certificación de embarque;
g) Se debe esperar para atender posibles preguntas del transportador para abrir los contenedores para inspeccionar o modificar el empaque.
En el laboratorio se debe asignar a una persona para recibir la muestra y hacer la custodia de la misma, también debe inspeccionar el estado y los sellos de la muestra, comparar la información que viene en el sello del recipiente de la muestra contra la información de la cadena de custodia, asignarle el número de radicación y colocarla en el cuarto frío hasta que sea programada para el análisis.
2. PROTOCOLOS METODOLOGICOS PARA CORROSIVIDAD
Para la evaluación de esta característica, se seleccionaron cuatro (4) pruebas diferentes: determinación de pH, determinación de reserva ácido/álcali, evaluación de corrosividad en lámina de acero y aplicación de la prueba corrositex®.
El orden de ejecución de estos procedimientos se presenta en la Figura 2.5. En esta, se advierte que la primera prueba a realizar corresponde a la determinación del pH del residuo. Si los resultados de esta prueba se encuentran dentro de los límites presentados, se considera que el residuo es corrosivo y la evaluación de la propiedad finaliza. Si por el contrario, el pH determinado se encuentra en el rango que indica que el residuo evaluado no es corrosivo, se procede a realizar la determinación de la reserva ácido/álcali, cuyo resultado combinado con el pH determinado en la etapa anterior permitirá la catalogación del residuo. Si los resultados se ajustan a los criterios presentados en la Figura 2.1, la prueba finaliza y se considera que el residuo exhibe propiedades corrosivas. En caso que este resulte ser no corrosivo desde el punto de vista de la combinación pH-Reserva ácido/álcali, se procede a hallar la tasa de corrosión producida por el residuo sobre lámina de acero y finalmente en caso de que los resultados se encuentren por debajo del límite establecido a las condiciones de la prueba, se finaliza la evaluación aplicando la prueba Corrositex®, indicativo de la corrosión que puede causar el residuo al entrar en contacto con la piel humana. Si el resultado de los cuatro (4) protocolos ejecutados arroja que el residuo no puede ser catalogado como corrosivo, se finaliza la serie de pruebas y se concluye que el residuo bajo estudio no exhibe propiedades características de residuos que presentan la propiedad de corrosividad.
A continuación se presentan los protocolos seleccionados para la determinación de distintas características, cuya evaluación permitirá la catalogación de un residuo como corrosivo.
2.1 Método de prueba medición electrométrica de pH
I. ALCANCE Y APLICABILIDAD
-- Este método mide el pH de desechos acuosos y de múltiples fases en los cuales la fase acuosa constituya al menos el 20% del volumen total del desecho.
-- Este método no se puede aplicar a ácidos y bases concentradas, ni a ácidos y bases concentrados mezclados con sustancias inertes.
-- Se requiere que el desecho tenga un contenido mínimo de agua para medir el pH. El método es aplicable a sustancias en estado sólido [1].
II. PRINCIPIO DEL METODO
El pH de la muestra es determinada electrométricamente usando un electrodo de vidrio en combinación con un electrodo de referencia o un electrodo combinado.
III. INTERFERENCIAS
-- El electrodo de vidrio, en general, no está sujeto a inferencias de la solución de color, turbiedad, coloides, oxidantes, reductores, o salinidad moderada (<0,1 molar).
-- El error por sodio a niveles de pH > 10 puede ser reducido o eliminado usando un electrodo de bajo error de sodio.
-- Capas de material aceitoso o material particulado pueden disminuir o dañar la respuesta del electrodo. Estas capas pueden ser removidas por una suave limpieza o un enjuague con detergente, seguidas por un enjuague con agua destilada. Tratamiento adicional con ácido clorhídrico (1:10) puede ser necesario para remover cualquier película remanente.
-- La temperatura puede afectar la determinación de pH por dos causas. La primera es el cambio en la respuesta del electrodo a varias temperaturas. Esta interferencia debe ser controlada con un instrumento con compensación de temperatura o calibrando el equipo con su electrodo a la temperatura de la muestra. La segunda causa es el cambio del pH debido a los cambios en la muestra por cambios en la temperatura. Este error es dependiente de la muestra y no puede ser controlado.
IV. EQUIPOS Y MATERIALES
-- pHmetro.
-- Electrodo de vidrio.
-- Electrodo de referencia: Un electrodo de cloro plata-plata u otro electrodo de referencia de potencial constante [2].
-- Agitador magnético y barra de agitación cubierta en teflón.
-- Termómetro y/o sensor de temperatura para compensación automática.
V. REACTIVOS
Soluciones tampón de estándar secundario preparado de sales NIST o comprados. Estas soluciones vendidas comercialmente deben ser validadas por comparación con estándar NIST. Se requiere tener como mínimo un buffer de pH 2 para desechos ácidos y un buffer pH 12 para desechos cáusticos.
VI. PROCEDIMIENTO
Calibración.
Se debe calibrar de acuerdo al procedimiento de operación del equipo. Cada sistema pHmetro/electrodo debe ser calibrado con un mínimo de dos (2) puntos que abarque el pH esperado de la muestra y su diferencia debe ser 3 o más unidades de pH. La medición para desechos con un valor de pH > 12 debe medirse a 25 ± 1 ºC. El ajuste se debe repetir en porciones sucesivas de las dos (2) soluciones buffer hasta que las lecturas estén dentro de 0,05 unidades de pH del valor de la solución buffer.
Procedimiento.
-- Localizar la muestra en un beaker de vidrio limpio, usando suficiente volumen para cubrir los elementos sensores del electrodo y dar un adecuado espacio para la barra de agitación magnética. Si se están realizando mediciones de campo, el electrodo puede ser inmerso directamente en la corriente de la muestra a una profundidad adecuada y se debe mover de manera que se asegure suficiente movimiento de la muestra a través de los elementos sensores del electrodo.
-- Si la temperatura de la muestra difiere en más de 2 ºC de la solución buffer, la medición de pH debe ser corregida.
-- Leer y registrar el pH de la muestra y la temperatura. Repetir la medida sobre alícuotas sucesivas de muestra hasta que los valores difieran por menos de 0,1 unidades de pH. Usualmente con 2 o 3 cambios es suficiente.
VII. RESULTADOS
La sustancia se podrá considerar corrosiva si el valor de lectura de pH se encuentra fuera de los rangos sugeridos por la normatividad.
VIII. INFORMACION ESTADISTICA
En la Tabla 2.1 se presentan los datos de exactitud de un estudio de desempeño con datos de cuarenta y cuatro (44) analistas, en veinte (20) laboratorios analizando, seis (6) muestras de agua sintética conteniendo incrementos exactos del ion hidróxilo.
Tabla 2.1
Información estadística determinación electrométrica de pH
| pH | Desviación estándar | Sesgo (%) | Sesgo (pH) |
| 3,5 | 0,10 | -0,29 | -0,01 |
| 3,5 | 0,11 | -0,00 | |
| 7,1 | 0,20 | +1,01 | +0,07 |
| 7,2 | 0,18 | -0,03 | -0,002 |
| 8,0 | 0,13 | -0,12 | -0,01 |
| 8,0 | 0,12 | +0,16 | +0,01 |
IX. REFERENCIAS
Método 9040C. Medición de pH electrométrica. SW 846. Revisión 3 noviembre 2004
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 2.1. Diagrama de flujo procedimiento para determinación electrométrica de pH.
2.2 Determinación reserva Acido/Alcali
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias sólidas y líquidas.
-- Permite determinar la reserva ácido/álcali de una sustancia, definida como la cantidad equivalente de base, necesaria para llevar el pH de esta hasta un valor previamente establecido.
II. PRINCIPIO DEL METODO
Una muestra de la sustancia en estado sólido o líquido es titulada con solución de hidróxido de sodio (sustancias con pH inferior a 4.0) o solución de ácido sulfúrico (sustancias con pH superior a 10.0) hasta valores de pH establecidos (4.0 y 10.0 para sustancias ácidas y alcalinas respectivamente). El valor de la reserva ácido álcali se expresa en términos de la cantidad equivalente de hidróxido de sodio necesaria para la titulación de cien (100) gramos de sustancia y constituye un indicativo de la capacidad buffer de la sustancia o residuo.
III. INTERFERENCIAS
No se reportan posibles interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
-- Bureta
-- Vaso de precipitado
-- Balanza analítica
-- pH metro
-- Agua destilada
V. REACTIVOS
-- Solución de ácido sulfúrico 2 N
-- Solución de hidróxido de sodio 2 N
VI. PROCEDIMIENTO
-- En caso de sustancias sólidas, tomar una muestra de aproximadamente 5 g en un vaso de precipitado. En caso de sustancias en estado líquido, tomar una muestra de aproximadamente 50 ml.
-- Registrar el peso exacto de la muestra.
-- Para muestras sólidas, disolver o suspender en 50 ml de agua destilada.
-- Registrar el pH de la muestra.
-- Si el pH de esta se encuentra por debajo de 4.0, llenar la bureta con la solución 2 N de hidróxido de sodio. Si el pH registra un valor superior a 10.0, llenar la bureta con la solución 2 N de ácido sulfúrico.
-- Para muestras con pH inferior a 4.0, agregar la solución de base hasta alcanzar un pH de 4.0 unidades.
-- Para muestras con pH superior a 10.0 agregar la solución de ácido a la muestra hasta alcanzar un pH de 10.0 unidades.
-- Registrar el volumen de solución consumido.
VII. RESULTADOS
La reserva ácido/álcali, expresada como g NaOH/ 100 g muestra, se calcula como:
8000*V
Reserva ácido/álcali =
m
Donde,
V = volumen de solución de NaOH o H2SO4 adicionado para alcanzar pH = 4.0 o pH= 10.0 (L).
m = peso de la muestra (g).
La definición de un residuo como corrosivo con base en la reserva ácido/álcali se realiza en combinación con el pH de este mediante el cálculo de:
pH + 1/12 reserva ácido/álcali y pH-1/12 reserva ácido/álcali
Si el mayor valor es igual o superior a 14.5 o el menor valor es igual o inferior a 0.5, el residuo se considera corrosivo
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 2.2. Diagrama de flujo procedimiento para determinación de reserva ácido/álcali.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
Young J, How M, Walker A, Worth W. Classification as corrosive or irritant to skin of preparations containing acidic or alkaline substances. Toxicology in Vitro. Vol 2: 19-26. 1988.
2.3 Método de prueba corrosión al acero
I. ALCANCE Y APLICABILIDAD
El método mide la corrosividad al acero de desechos líquidos acuosos y no acuosos. Con base en los resultados obtenidos se puede identificar si el desecho presenta la característica de corrosividad como un peligro físico.
II. PRINCIPIO DEL METODO
Este método expone cupones de tipo Acero SAE 1020 al desecho líquido que se va a evaluar en una relación mínima de volumen del desecho a área del cupón metálico de 40 ml/cm2, a una temperatura de 55 ºC y tiempo determinados, después de los cuales se mide el grado de pérdida de material del cupón. Se determina la corrosividad de dicho desecho por diferencia de peso del cupón.
III. INTERFERENCIAS
Se pueden presentar grandes diferencias ocasionadas cuando la superficie del metal se ha pasivado. Se pueden presentar resultados subestimados cuando hay pérdidas de masa en puntos localizados como grietas.
IV. EQUIPOS Y MATERIALES
-- Se debe usar un recipiente de tamaño adecuado (usualmente entre 500 a 5.000 ml) con tres (3) cuellos de dimensión apropiada (por ejemplo NS29/32), uno de ellos para el condensador de reflujo. Este debe ser construido de un material que no sea afectado por el residuo y que no contamine el residuo que se esta evaluando (por ejemplo, vidrio o PTFE). Un recipiente típico para este tipo de ensayo se muestra en la figura 2.3.
-- Termómetro y mecanismo de regulación de temperatura.
-- Aparato de calentamiento (manta, plancha o baño).
-- Sistema de soporte para los especímenes metálicos. El método de soporte de los cupones puede variar con el aparato usado para realizar el ensayo, sin embargo este debe ser diseñado para que los especímenes estén aislados física y eléctricamente uno de otro, y para que estos permanezcan aislados de cualquier elemento metálico u otro elemento usado en el ensayo. Este debe ser construido de un material que no sea afectado por el residuo y que no contamine el residuo que se está evaluando algunos materiales de soporte son vidrio, hilos de PTFE no extruido, fluorocarbonados o metal revestido.
-- Cupones o especímenes fabricados en acero SAE 1020. El tamaño y forma debe asegurar el contacto libre con el desecho. Todos deben ser medidos rigurosamente con calibrador métrico a una exactitud de 0,01 mm para permitir un cálculo exacto del área expuesta. Una forma conveniente es un espécimen circular de alrededor de 3.75 cm de diámetro con un espesor aproximadamente de 0.32 cm. El espécimen debe tener un orificio para colocarse en la montura de soporte, cuyo diámetro aproximado puede ser de 0.80 cm. El área superficial total de un espécimen circular estará dado por la siguiente ecuación:
![]()
Donde:
t | espesor del espécimen |
d | diámetro de hoyo para colgarlo en el soporte |
D | diámetro del espécimen |
Si el hoyo del espécimen es cubierto completamente por la montura del soporte, el último término de la ecuación se omite. Se pueden esperar resultados más uniformes si una capa sustancial del metal es removida de los cupones antes del ensayo.
-- Balanza analítica con precisión de ± 0,001 g o mejor.
-- Mecanismo de agitación.
-- Lija abrasiva No 120
-- Desecador
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 2.3. Equipo para medida de corrosividad sobre acero SAE 1020.
A: termómetro.
B: Frasco de cristal.
C: especímenes metálicos colgados en el dispositivo de soporte
D: manta caliente (placa de calentamiento).
E: interfase líquida.
F: abertura de la tapa para añadir aparatos adicionales de ser necesarios.
G: condensador de reflujo.
V. REACTIVOS
Solución de limpieza química 1.
Solución de hidróxido de sodio al 20% con 200 g/L de polvo de zinc.
-- Hidróxido de sodio (NaOH) (20%): disolver 200g de NaOH en 800 ml agua tipo II y mezclar bien.
-- Polvo de Zinc grado reactivo.
Solución de limpieza química 2.
HCl concentrado con 50 g/l de cloruro estañoso y 20 g/l de cloruro de antimonio.
-- Acido Clorhídrico (HCl) concentrado.
-- Cloruro estañoso (SnCl2).
-- Cloruro de antimonio (SbCl3).
Solvente (requerido para limpieza química, uno de las siguientes alternativas).
-- Acetona.
-- Diclorometano.
-- Alcohol.
-- Acetona o Metanol.
-- Detergente sin blanqueador.
-- Agua destilada.
-- Acido sulfúrico (requerido si se va a realizar limpieza electrolítica).
-- Eter diortotolil tiourea (Inhibidor que se puede utilizar para la limpieza electrolítica).
VI. PROCEDIMIENTO
Limpieza inicial.
Pulir los cupones o especímenes por tratamiento químico, remoción electrolítica, o pulido con abrasivo grueso. Al menos se debe remover 0,254 mm o 2 a 3 mg/cm2. El tratamiento final de la superficie del espécimen incluye utilizar papel o tela abrasiva (lija) No 120. La limpieza final consiste en restregar con blanqueador libre de polvo, seguido de un enjuague con agua destilada y luego en acetona o metanol, por último secar al aire. Al final de la limpieza el cupón debe colocarse en el desecador hasta ser utilizado. Cada espécimen o cupón debe ser estampado, grabado, cortado con una marca de identificación adecuada, esto puede generar corrosión localizada.
-- Pesar los especímenes metálicos.
-- Ensamblar el montaje del recipiente, con termómetro, aparato de calentamiento, equipo de agitación. Colocar los cupones en el sistema de soporte. Se deben utilizar tres (3) cupones en el ensayo; uno totalmente sumergido con una distancia entre el borde superior del cupon y la superficie del líquido de 10 mm, otro sumergido la mitad en el líquido, y el tercero en la fase gaseosa.
-- Llenar el contenedor con la cantidad apropiada de desecho, considerando que se debe mantener la relación mínima de volumen del desecho a área del cupón metálico de 40 ml/cm2.
-- Comenzar la agitación a una velocidad que asegure que el líquido se mantenga bien mezclado y homogéneo. En residuos que tengan su punto de ebullición cerca de la temperatura del ensayo, la convección térmica puede ser una adecuada fuente de agitación por lo cual no se requeriría de ningún equipo extra. En soluciones con altas viscosidades, se recomienda el suministro de agitación.
-- Utilizando el dispositivo de calentamiento llevar la temperatura del residuo a 55 ºC ± 1 ºC. Las pérdidas por evaporación deben ser controladas por un mecanismo de nivel constante o por adiciones frecuentes del residuo para mantener el volumen original dentro de un ± 5%.
-- Mantener las condiciones durante 24 horas, al final de los cuales se retiran los cupones.
Limpieza final de los cupones.
El procedimiento de limpieza debe remover todos los productos de la corrosión removiendo un mínimo del metal sano. La limpieza puede realizarse por tres métodos diferentes: mecánico, químico y electrolítico. Después de esta limpieza, el cupón se debe restregar con blanqueador libre de polvo, seguido de un enjuague con agua destilada y luego enjuagar con abundante solvente (acetona o metanol). Por último secar al aire. Al final de la limpieza el cupón debe colocarse en el desecador hasta ser utilizado. Sea cual sea el tratamiento que se use para limpiar los especímenes, su efecto en la remoción de metal sano debe ser determinada por medio de un blanco (un espécimen que no haya sido expuesto al residuo). El blanco se debe pesar, luego limpiar junto con el espécimen de la prueba, y al final pesar.
-- Limpieza mecánica: Incluye restregar, lijar, cepillar y procedimientos ultrasónicos. El método más utilizado es restregar con un cepillo de cerdas y un abrasivo suave, los otros se usan en casos de alta corrosión, como primer paso antes de restregar. Se debe tener precaución de no remover metal sano.
-- Limpieza química: Remoción de material en la superficie del especímen por dilución en un solvente apropiado y luego inmersión en la solución de limpieza. Solventes como la acetona, diclorometano, y alcohol, se utilizan para remover aceites, grasas, o materiales resinosos.
Las soluciones convenientes para eliminar la corrosión del acero son:
| Soluciones de limpieza | Tiempo de inmersión | Temperatura |
20% NaOH + 200 g/l polvo Zinc | 5 minutos | Ebullición |
HCl conc.+ 50 g/l SnCl2+ 20 g/l SbCl3 | hasta estar limpio | Frío |
-- Limpieza electrolítica: esta debe ser precedida por un cepillado para remover los productos adheridos de la corrosión. A continuación se presenta un método de limpieza electrolítico que puede ser usado:
| Solución | 50 g/l H2SO4 |
| Anodo | carbón o plomo |
| Cátodo | espécimen de acero |
| Densidad actual del cátodo | 20 amp/cm2 |
| Inhibidor | 2 cc inhibidor orgánico/l |
| Temperatura | 74 ºC |
| Período de exposición | 3 min. |
Se debe tener precaución de asegurar un buen contacto eléctrico con el cupon para evitar contaminación de la solución de limpieza con iones metálicos fácilmente reducibles y asegurar que no ocurra descomposición del inhibidor. Como inhibidores se puede usar 0,5 g/l de éter diortotolil tiourea.
-- Pesar los especímenes. La pérdida de peso se usa como la principal medida de la corrosión. Para poder utilizar esta pérdida de peso como una medida de corrosión, se requiere asumir que toda la pérdida de peso ha sido causada por la corrosión y no por pérdidas locales.
VII. RESULTADOS
Para determinar la tasa de corrosión se usa la siguiente fórmula:

Donde:
Wic: | peso inicial del cupón después de la limpieza inicial (mg). |
Wfc: | peso final del cupón después de la exposición a la sustancia en evaluación y del proceso de limpieza realizado (mg). |
Wib: | peso inicial del cupón usado como blanco antes del proceso de limpieza (el mismo realizado para el cupón del ensayo) (mg). |
Wfb: | peso final del cupón usado como blanco después del procesos de limpieza (el mismo realizado para el cupón del ensayo) (mg). |
A: | área (cm2). |
T: | tiempo (horas). |
11,145: factor de conversión considerando la densidad del acero tipo SAE 1020 7,86 g/cm3, y convertir unidades a tasa de corrosión en mm/año.
-- Se debe reportar la forma y dimensiones del cupón o espécimen y el método usado para preparar los especimenes para el ensayo.
-- Se debe reportar el método de limpieza después de la exposición.
-- En los casos en los cuales la corrosión no pudo ser removida totalmente, la perdida de masa debe ser reportada, pero con una observación indicando que algunos productos de la corrosión no fueron removidos.
-- Tasas de corrosión para cada espécimen evaluado.
-- Se usa el valor del espécimen con mayor tasa de corrosión.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
-- Method 1110A Corrosivity Toward Steel SW-846.
-- Sección 37. Procedimientos de clasificación, métodos de prueba y criterios relativos a las sustancias de la clase 8. Libro de Pruebas y Criterios – Naranja de las Naciones Unidas.
NACE Internacional the corrosion society. Estándar Test Method Laboratory Corrosion Testing of Metals NACE Standard TM0169-2000 item No 21200
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 2.4. Diagrama de flujo procedimiento para determinación de corrosión al acero.
2.4 Diagrama metodológico determinación característica de corrosividad
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 2.5. Arbol de decisión para ejecución de pruebas relativas a clasificación de residuos corrosivos.
3. Protocolos metodológicos para explosividad
La evaluación de la explosividad de un residuo, involucra diferentes pruebas de tipo complementario, que determinan desde diferentes ángulos si un residuo puede ser catalogado como tal.
Estas pruebas incluyen la determinación de la capacidad de propagación de la detonación, la sensibilidad ante condiciones de calor intenso, el efecto de la inflamación en espacio limitado y la sensibilidad a estímulos mecánicos, específicamente choque y fricción.
Un resultado positivo en cualquiera de estos ensayos indica que el residuo es explosivo, sin que un resultado negativo signifique que el residuo pueda ser considerado como no explosivo, debido a que cada prueba evalúa reacciones ante estímulos específicos y por tanto pueden presentarse estímulos diferentes ante los cuales el residuo exhiba características explosivas.
El orden de ejecución de las distintas pruebas no es específico, debido a que como se mencionó anteriormente, se trata de pruebas complementarias. Sin embargo, debido a la complejidad de algunos de los procedimientos, se plantea una secuencia que inicia el proceso de evaluación de esta característica mediante la aplicación de las pruebas con el menor grado de dificultad en su realización (Figura 3.17).
A continuación se presentan los cinco (5) protocolos seleccionados para la determinación de distintas características, cuya evaluación permitirá la catalogación de un residuo como explosivo.
3.1 Prueba para determinar la propagación de la detonación-prueba de excitación con barrera interpuesta
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias líquidas y sólidas.
-- Permite determinar si una sustancia en condiciones de espacio limitado, al ser sometida a la detonación producida por una carga multiplicadora, tiene la capacidad de propagar dicha detonación.
II. PRINCIPIO DEL METODO
Una sustancia en estado líquido o sólido es introducida en un tubo de acero de dimensiones específicas y puesta en contacto con un material detonante con el fin de determinar si la sustancia propaga la detonación producida. En este caso, la sustancia se considera como explosiva.
III. INTERFERENCIAS
No se reportan posibles interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
-- Equipo compuesto por un tubo de acero al carbono, estirado en frío y sin costuras, con un diámetro exterior de 48 ± 2 mm, un espesor de pared de 4.0 ± 0.1 mm y una longitud de 400 ± 5 mm. En caso que la sustancia bajo prueba pueda reaccionar con el acero, el tubo debe ser revestido interiormente con resina de fluorocarbono. El extremo inferior del tubo se encuentra cerrado por dos capas de polietileno laminado de 0.08 mm de espesor, sujetas mediante tiras de goma elástica y cinta aislante de tal manera que se permita la deformación plástica. En caso que la sustancia bajo prueba presente reacción con el polietileno, este material puede ser reemplazado por láminas de politetrafluoretileno. En el extremo superior del tubo se encuentra una placa de acero suave de 150 ± 10 mm de lado y 3.2 ± 0.2 mm de espesor, que está separada del tubo por dos elementos de 1.6 ± 0.2 mm de espesor y que se conoce como placa testigo. La Figura 3.1 presenta un esquema del equipo y sus partes constitutivas.
Detonador.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.1. Equipo para prueba de excitación con barrera interpuesta.
V. REACTIVOS
Carga multiplicadora de ciclonita/cera (95/5) o de pentrita/TNT (50/50) de 50 ± 1 mm de diámetro con una densidad de 1600 ± 50 kg/cm3 (longitud de 50 mm).
VI. PROCEDIMIENTO
-- Se llena el tubo de acero con la sustancia a estudiar. En el caso de sustancias sólidas, se introduce una cantidad suficiente para llenar el tubo y este se golpea muy suavemente hasta que la sustancia se comprima en su interior; posteriormente se agrega una cantidad adicional de muestra y se repite el procedimiento hasta que nuevamente se complete la totalidad del volumen del tubo.
-- Se coloca el tubo en posición vertical.
-- Se toman 160 g de la carga multiplicadora.
-- Si se trata de ciclonita/cera, esta puede prensarse en una o varias piezas mientras la carga total corresponda a las especificaciones. Si se trata de pentrita/TNT esta debe ser moldeada.
-- La carga multiplicadora se pone en contacto directo con la lámina que sella el extremo inferior del tubo.
-- Se coloca el detonador contra la carga multiplicadora y se activa. Se realizan dos ensayos en caso de no presentarse detonación en el primero.
VII. RESULTADOS
Se considera que la sustancia puede propagar la detonación y por tanto es explosiva, si en la prueba el tubo se fragmenta completamente o la placa testigo resulta perforada. Si no se presenta ninguno de estos resultados, se considera que la sustancia no propaga la detonación, más no que la sustancia no sea explosiva. En la Tabla 3.1 se presentan algunos resultados obtenidos con diferentes sustancias estudiadas.
Tabla 3.1
Resultados obtenidos con diferentes sustancias aplicando la prueba de excitación con barrera interpuesta
| Sustancia | Densidad aparente (kg/m3) | Longitud de Fragmentación (cm) | Placa testigo | Resultado |
| Nitrato amónico | 800 | 40 | Abovedada | Positivo |
| Nitrometano | 1130 | 40 | Perforada | Positivo |
| Perclorato amónico | 1190 | 40 | Perforada | Positivo |
| Agua | 1000 | <40 | Abovedada | Negativo |
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.2. Diagrama de flujo procedimiento para determinación de sustancias que tienen la capacidad de propagar la detonación.
IX. REFERENCIAS
UNECE-Manual de Pruebas y Criterios-Prueba 1 a).
3.2 Prueba de sensibilidad ante condiciones de calor intenso-prueba koenen
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias líquidas y sólidas.
-- Permite determinar la sensibilidad de sustancias en estos estados ante los efectos de calor intenso en un espacio muy limitado.
II. PRINCIPIO DEL METODO
En el método, la sustancia sólida o líquida se introduce en un tubo en condiciones específicas y se somete a calentamiento mediante gas propano, realizando el sellado del tubo con placas que presentan un orificio de distintos diámetros. La prueba se realiza iniciando con la placa de mayor diámetro (20.0 mm) y en caso de que no se presente explosión se utilizan las placas con orificios de menor diámetro hasta que se presente explosión, teniendo como límite un diámetro de 1.0 mm. En caso que no se presente explosión utilizando estos diámetros, o se observen determinados efectos en el sistema al finalizar la prueba, la sustancia se considera no sensible al calentamiento en espacio confinado. En caso que se presente explosión, se analiza el tipo de fragmentación obtenido y se determina si la sustancia puede ser considerada como explosiva.
III. INTERFERENCIAS
No se reportan posibles interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
-- El equipo necesario para la prueba está constituido por un tubo de lámina fina de acero no reutilizable, con un sistema de cierre reutilizable. El tubo, cuyas dimensiones y características se presentan en la Figura 3.3, se encuentra ubicado en un dispositivo de calentamiento y protección y tiene un peso de 25.5± 1 g. En el extremo abierto del tubo se encuentra una brida cubierta con una tuerca ajustable con llave 36 y un collar roscado. La placa de cierre a través de la cual salen los gases resultantes de la descomposición está elaborada en acero al cromo, resistente al calor y el diámetro del orificio de salida se encuentra en medidas entre 20 y 1 mm. Finalmente se encuentra del dispositivo de cierre del equipo, formado por una tuerca de diámetro 10.0 o 20.0 mm. ajustable con llave 41.
-- El dispositivo de calentamiento y protección en el que se introduce el tubo cuenta con dos varillas apoyadas en orificios que se encuentran en paredes opuestas de la caja. Entre estas varillas se suspende el tubo al realizar la prueba. Igualmente en el dispositivo se encuentran cuatro (4) quemadores con sistema de encendido eléctrico. La distribución de los quemadores en el sistema se presenta en la Figura 3.4.
-- Gas propano provisto en cilindro con regulador de presión.
-- Termopar de 1 mm. de diámetro.
-- Sistema de extracción de gases.
-- Lubricante a base de disulfuro de molibdeno.
V. REACTIVOS
-- Dibutilftalato
VI. PROCEDIMIENTO
Primera etapa.
-- Se introducen 27 cm3 de dibutilftalato en el tubo del equipo.
-- Se sella el tubo con una placa de cierre de 1.5 mm de diámetro.
-- Se ubica el tubo en la caja de calentamiento y protección.
-- Se ubica un termopar en el centro del tubo 43 mm por debajo del borde.
-- Se evacua la zona de la prueba.
-- Se permite el paso de gas propano y su distribución a través del sistema de quemadores.
-- Se registra el tiempo necesario para que la temperatura del líquido pase de 135 a 285 ºC.
-- Se calcula la velocidad de calentamiento mediante:
Donde:
V | velocidad de calentamiento (K/s) |
t | tiempo empleado para aumentar la temperatura de 135 a 285 ºC (s) |
-- Se repite el procedimiento, regulando la presión del gas hasta que la velocidad de calentamiento resulte ser de 3.3 ± 0.3 K/s.
Segunda etapa.
-- En el caso de sustancias sólidas, se introducen en un tubo 9 cm3 de sustancia y se compactan con una fuerza de 80 N aplicada a la sección transversal del tubo.
-- Si el material es compresible, se adiciona una nueva cantidad de la sustancia y se compacta hasta que la muestra en el tubo llega hasta 55 mm del borde.
-- Se determina la masa total adicionada hasta ese nivel.
-- En una etapa posterior, se toma un nuevo tubo y se adiciona una cantidad de muestra compactada igual a la tercera parte de la masa determinada anteriormente.
-- Se agregan dos muestras adi
cionales de la sustancia previamente compactadas a 80N.
-- Se adiciona una nueva cantidad de muestra compactada o se retira hasta que la sustancia en el tubo se encuentre a 15 mm del borde de este.
-- La cantidad de muestra empleada en esta etapa corresponde a la que será utilizada en cada prueba.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.3. Equipo empleado para prueba Koenen.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.4. Dispositivo de calentamiento y protección-Prueba Koenen.
Tercera etapa.
-- Se efectúa el llenado del tubo introduciendo la muestra en tres (3) porciones iguales, cada una comprimida hasta un volumen de 9 cm3. -- En el caso de sustancia líquidas y geles, la muestra se introduce en el tubo hasta 60 mm del borde de este, cuidando que en el caso de geles no queden espacios vacíos.
-- Se realiza el montaje del collar roscado introduciéndolo desde la parte inferior del tubo y se inserta la placa de orificio de 20 mm de diámetro.
-- Se revisa que no haya restos de sustancia entre la placa y la brida o en las roscas.
-- Se ubica la tuerca de cierre de diámetro correspondiente[3] y se ajusta de manera manual después de aplicarle un lubricante a base de disulfuro de molibdeno.
-- Se coloca el tubo en una prensa de banco y se aprieta la tuerca de cierre con una llave.
-- Se ubica el tubo en el dispositivo de calentamiento y protección mediante el uso de las dos varillas ubicadas en este.
-- Se evacua la zona de prueba.
-- Se abre el suministro de gas, regulando la presión al nivel hallado en la primera etapa.
-- Si no estalla el tubo, se prolonga el calentamiento durante cinco (5) minutos como mínimo antes de dar por terminada la prueba.
-- En caso de explosión, se recogen y pesan los fragmentos del tubo.
-- En caso que no se presente explosión, se realiza nuevamente el procedimiento utilizando las placas con orificio de 12.0, 8.0, 5.0, 2.0, 1.5 y 1.0 mm de diámetro hasta que se presente explosión con alguna de esta aberturas.
VII. RESULTADOS
Se considera que la sustancia no reacciona ante el calentamiento en espacio confinado, mas no que es una sustancia no explosiva, si en las pruebas realizadas no se presenta explosión o se observa alguno de los siguientes efectos (Figura 3.6): “O”: tubo intacto, “A”: curvatura del fondo del tubo, “B”: curvatura del fondo y la pared del tubo, “C”: fractura del fondo, “D”: fractura de la pared, “E”: rompimiento del tubo en dos fragmentos.
Se considera que la sustancia es explosiva si al realizar la prueba se observa (Figura 3.6):
-- “F”: fragmentación del tubo en tres (3) o más partes, en su mayoría grandes, que en algunos casos pueden estar unidos entre sí por una tira estrecha.
-- “G”: fragmentación del tubo en diversos trozos, en su mayoría pequeños y dispositivo de cierre intacto.
-- “H”: fragmentación del tubo en diversos trozos muy pequeños, curvatura o fragmentación del dispositivo de cierre.
En la Tabla 3.2 se presentan algunos resultados obtenidos con diferentes sustancias estudiadas.
Tabla 3.2
Resultados obtenidos con diferentes sustancias aplicando la prueba de excitación con barrera interpuesta
| Sustancia | Diámetro límite de orificio de la placa al cual se presenta explosión (mm.) | Resultado |
| 1,3-dinitrobenceno | < 1.0 mm | Negativo |
| Nitrometano | < 1.0 mm | Negativo |
| Nitrato amónico | 1.0 mm | Positivo |
| Perclorato de amonio | 3.0 mm | Positivo |
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.5. Diagrama de flujo procedimiento para determinación de la sensibilidad de sustancias ante el calentamiento en espacio limitado.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.6. Posibles efectos en el tubo utilizado en el procedimiento-Prueba Koenen.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
UNECE-Manual de Pruebas y Criterios-Prueba 1 b).
3.3 Prueba para determinar el efecto de la inflamación en espacio limitado-prueba de tiempo/presión.
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias en estado sólido y líquido.
-- Permite determinar los efectos que causaría la ignición de la sustancia en espacios limitados con el fin de establecer si la ignición puede provocar una deflagración de violencia explosiva.
II. PRINCIPIO DEL METODO
En este método una muestra sólida o líquida de la sustancia bajo estudio es sometida a ignición en un recipiente confinado, al cual fue adaptado un sistema de ignición y de registro de presión, y tiene como objeto registrar el tiempo necesario para que la presión aumente de 690 KPa a 2070 Kpa[4].
III. INTERFERENCIAS
No se reportan interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
-- El equipo utilizado en la realización de prueba se presenta en la Figura 3.7. Está constituido por un recipiente cilíndrico en acero a presión, de 89 mm de longitud y 60 mm de diámetro exterior, el cual en cada uno de los extremos tiene una reducción maquinada a diámetros de 50 mm, en donde respectivamente se ajustan un tapón de activación para la ignición y una salida de gases. El aparato cuenta con una toma de presión ubicada en su superficie curva a 35 mm de uno de los extremos del tubo formando un ángulo de 90o con las superficies del tubo, y con una salida de 55mm del cuerpo del recipiente y 6 mm de diámetro interior de toma. El extremo del recipiente más alejado a la toma de la presión está cerrado con un tapón de activación que tiene dos electrodos, uno de los cuales va aislado del tapón y el otro está puesto a tierra en este último. El otro extremo del recipiente va cerrado por una cápsula de seguridad, fabricada en aluminio de 0,2 mm de espesor con una presión de rotura de 2200 KPa, y que se mantiene fija mediante un tapón que tiene un orificio de 20 mm de diámetro.
-- El equipo se ubica en un soporte (Figura 3.8) que mantiene en posición correcta el recipiente cilíndrico compuesto de una placa de acero de 235 mm x 184 mm x 6mm y de un tubo hueco de sección cuadrada de 70 mm x 70 mm x 4 mm y de 185 mm de longitud.
-- Dispositivo de ignición (Figuras 3.9 y 3.10) compuesto por una cabeza de encendido eléctrico, del tipo que se utiliza en los detonadores de baja tensión y de un trozo cuadrado de lienzo cebado (tela de lino recubierta por ambos lados de una composición pirotécnica de nitrato potásico, silicio y pólvora negra sin azufre) de 13 mm de lado.
-- Dispositivo para la medición de presión que debe resistir los gases calientes y los productos de descomposición de la sustancia en análisis, y cuya sensibilidad permita registrar aumentos de presión de 690 a 2070 KPa en menos de 5 minutos.
V. REACTIVOS
No se requieren.
VI. PROCEDIMIENTO
-- Para sustancias sólidas se separan los contactos planos de latón de la cabeza de encendido del aislador. Luego se corta la parte descubierta del aislador. Posteriormente la cabeza de encendido se une al tapón de activación, soldándose los bornes de este con los contactos de latón, de manera que la parte superior de la cabeza de encendido sobresalga 13 mm de la cara superior del tapón de activación. Se perfora en el centro un trozo de lienzo cebado
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.7. Equipo para realización de prueba tiempo/presión.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.8. Soporte equipo para realización de prueba tiempo/presión.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.9. Sistema de ignición para sólidos equipo para realización de prueba tiempo/presión.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.10. Sistema de ignición para líquidos equipo para realización de prueba tiempo/presión.
cuadrado de 13 mm de lado; este se coloca sobre la cabeza de encendido y se enrolla sobre está sujetándola con un hilo fino de algodón.
-- Para sustancias líquidas, los cables se sueldan con los contactos de la cabeza de encendido. De manera posterior, se introducen los cables en el interior de un trozo de 8 mm de longitud de tubo de caucho de silicona de 5 mm de diámetro y 1 mm de diámetro interior. A continuación se hace subir el tubo hasta los contactos de la cabeza de encendido. Posteriormente, el trozo de lienzo cebado se enrolla sobre la cabeza y se utiliza una funda de cloruro de polivinilo o material equivalente para recubrir el lienzo cebado y el tubo de caucho de silicona. La funda se fija herméticamente enrollando un alambre fino sobre el tubo y la funda. Luego, se sueldan los cables a los bornes del tapón de activación de tal manera que la parte superior de la cabeza de encendido sobresalga 13 mm sobre la cara superior del tapón.
-- Una vez montado el aparato con el medidor de presión pero sin la cápsula de seguridad y sujetándolo con el tapón de activación hacia abajo, se introduce la muestra de 5 gramos de la sustancia a valorar de tal forma que esté en contacto con el dispositivo de ignición.
-- Posteriormente se coloca la arandela de seguridad y se enrosca al máximo el tapón de ventilación. Se monta el recipiente ubicado de manera que la cápsula de seguridad quede arriba en el soporte de ensayo, que se aloja por razones de seguridad en una campana blindada o en un cubículo a propósito.
-- Se conecta un explosor de dínamo a los bornes exteriores del tapón de activación y se procede a la realización de esta.
-- La señal producida por el transductor de presión se capta mediante el dispositivo para analizar la curva tiempo/presión y obtener un registro permanente de esta.
-- El ensayo se realiza tres (3) veces. Se anota el tiempo que tarda la presión en cambiar de 690 KPa a 2070 KPa por encima de la presión atmosférica. Se considera el valor más pequeño.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.11. Diagrama de flujo procedimiento para prueba tiempo/presión.
VII. RESULTADOS
Se considera que el resultado es positivo y que la sustancia es capaz de deflagrar si la presión máxima alcanzada en la prueba es igual o superior a 2070 KPa. Se considera que el resultado es negativo y que no es probable que se produzca deflagración si la presión alcanzada en cualquiera de los ensayos es inferior a 2070 KPa.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
UNECE-Manual de Pruebas y Criterios-Prueba N.4.
3.4 Prueba para determinar el efecto de la inflamación en espacio limitado-prueba de inflamación interior
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias sólidas y sustancias en estado coloidal.
-- Permite determinar la tendencia de una sustancia a experimentar una transición de deflagración a detonación.
II. PRINCIPIO DEL METODO
En este método, una sustancia en estado sólido o coloidal es introducida en un ambiente confinado (tubo de acero) y puesta en contacto con un inflamador de pólvora negra, activado mediante corriente eléctrica. Se evalúa el tipo de fragmentación que sufre el tubo con el fin de determinar si la sustancia es explosiva, o no presenta tendencia a experimentar una transición de deflagración a detonación.
III. INTERFERENCIAS
No se reportan interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
-- El equipo utilizado en el ensayo (Figura 3.12), consta de un tubo de acero al carbono (A53, calidad B) de 3 pulgadas, ficha 80”, con diámetro interno de 74 mm y 7.6 mm de espesor de pared. Los extremos del tubo se encuentran cerrados con un tapón hembra roscado de acero forjado de 3000 lb. En el centro del tubo se ubica un inflamador de pólvora negra de tamaño de partícula menor a 0.297 mm (pasa tamiz No 20 de 0,297 mm). El inflamador consiste en un recipiente cilíndrico de 21 mm de diámetro y 64 mm de largo elaborado en acetato de celulosa de 0.54 mm de espesor. Este se mantiene fijo mediante dos capas de cinta del mismo material reforzadas con hilos de nylon. El recipiente contiene un circuito en bucle de 0.30 mm de diámetro, constituido por un hilo de 25 mm de longitud y resistencia de 0.35 O, fabricado en una aleación de cromo-níquel. El bucle se encuentra conectado a dos hilos conductores aislados de 0.7 mm de diámetro, cuyo material es cobre estañado. Los hilos sobresalen del tubo a través de dos orificios pequeños que se taponan con resina epóxica.
-- Transformador de 20 V
V. REACTIVOS
-- No se requieren
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.12. Equipo de prueba para determinación de inflamación interior.
VI. PROCEDIMIENTO
-- Se introduce la muestra en el tubo hasta una altura de 23 cm.
-- Se dispone el inflamador en el centro del tubo y se hacen pasar los cables de este a través de los orificios en la pared.
-- Se tensionan los cables y se aíslan con resina epóxica.
-- Se introduce la parte restante de la muestra. En el caso de sustancias granulares, al introducir la muestra, se golpea suavemente en varias oportunidades el tubo de manera que se logre su compactación.
-- Se pone el tapón superior del tubo.
-- Se pone el tubo en posición vertical.
-- Se activa el inflamador con una corriente de 15 A procedente de un transformador de 20 V.
-- Se realizan tres (3) ensayos con cada muestra a no ser que la transición de deflagración a detonación se produzca en una prueba anterior.
VII. RESULTADOS
Se considera que la sustancia es explosiva si el tubo o uno de los tapones roscados se fragmentan en dos partes. Se considera que la sustancia no presenta tendencia a experimentar una transición de deflagración a detonación, mas no que la sustancia no sea explosiva, en caso que el tubo se abra o se agriete o cuando el tubo o los tapones se deformen hasta el punto que estos últimos se desprendan.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
UNECE-Manual de Pruebas y Criterios-Prueba 1 c) ii.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.13. Diagrama de flujo procedimiento para determinación del efecto de la inflamación en espacio limitado – prueba de inflamación interior.
3.5 Sensibilidad a estímulos mecánicos-choque y fricción
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias líquidas, sólidas y pastosas.
-- El método permite determinar si una sustancia en estos estados presenta peligro de explosión al ser sometida a estímulos mecánicos consistentes en choques y fricción para el caso de sólidos y choques en el caso de sustancias líquidas.
II. PRINCIPIO DEL METODO
El método determina la posibilidad de explosión de sólidos al ser sometidos al efecto de choque o fricción (sensibilidad mecánica), y el peligro de explosión de líquidos al ser sometidos a choques. La sensibilidad a los choques implica el sometimiento de la muestra al choque de un objeto de masa específica que cae desde una altura determinada. La sensibilidad a la fricción es determinada al someter a la muestra sólida o pastosa a fricción entre superficies estándar bajo condiciones específicas de carga y movimiento relativo.
III. INTERFERENCIAS
No se reportan interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
Sensibilidad mecánica-choque.
La Figura 3.14 (a) y (b), presenta las partes constitutivas y características del equipo necesario para la realización de la prueba. Este consta de bloques de acero ubicados en una base, yunques del mismo material, columnas, guías, pesos de 5 y 10 kg utilizados como cargas para la sustancia bajo estudio (Figura 3.14 (e)), un mecanismo de liberación y uno para el sostenimiento de la muestra. El yunque tiene 100 mm de diámetro y 70 mm de altura y se encuentra sujeto a la parte superior del bloque de acero mediante tornillos instalados para tal fin. El bloque de acero presenta las siguientes dimensiones: 230 mm de longitud, 250 mm de ancho y 200 mm de altura con un molde que sirve como base, con 450 mm de longitud, 450 mm de ancho y 60 mm de altura. Una columna construida en acero se encuentra sujeta a la parte inferior del bloque de acero y el equipo se encuentra ubicado en un bloque de concreto sólido de 60 x 60 x 60 cm. La sustancia es introducida en un mecanismo de choque, (2.14 (c) y (d)), que consta de dos (2) cilindros coaxiales fabricados en acero macizo pulido, con bordes redondeados, ubicados uno sobre el otro. El diámetro de los cilindros es de 0.003 a 0.005 mm y su altura es de 10 mm. Para el caso de sustancias sólidas, los cilindros se encuentran juntos, mientras que el mecanismo de choque para líquidos debe tener una separación de 1.0 mm entre los cilindros.
Sensibilidad mecánica-fricción.
El equipo utilizado para evaluar la sensibilidad a la fricción de una muestra, es presentado en la Figura 3.15. Está constituido por un cilindro de porcelana fijo y una placa móvil del mismo material. La placa de porcelana se encuentra sostenida en una carrocería que se desplaza sobre dos guías. Esta carrocería está conectada a un motor eléctrico de manera que la placa es movida solo una (1) vez hacia atrás y hacia delante una distancia de 10 mm. El cilindro de porcelana aplica generalmente fuerzas de 120 o 360 N. La placa de porcelana de 25 mm de longitud y 5 mm de altura y el cilindro, están elaborados de porcelana técnica blanca (rugosidad 9 a 32 ím). Las dimensiones del cilindro son 15 mm de largo y 10 mm de diámetro, con bordes redondeados de radio de curvatura igual a 10 mm.
V. REACTIVOS
No se requieren.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.14. Equipo de prueba para determinación de explosividad por sensibilidad al choque:
a) Vista frontal;
b) Vista de la parte inferior del equipo;
c) Detalles mecanismo de choque para sustancias sólidas y pastosas;
d) Detalles mecanismo de choque para sustancias líquidas;
e) Detalles martillo de caída (masa de choque).
VI. PROCEDIMIENTO
Sensibilidad mecánica-choque.
-- Tomar una muestra de 40 ml de la sustancia bajo prueba. En el caso de sustancias sólidas, someterlas a secado y tamizado para la selección de partículas que pasen tamiz de malla 0.5 mm. Si la sustancia se encuentra en gránulos es necesaria su pulverización y posterior tamizado. Para sustancias pastosas se debe realizar un procedimiento de secado, pulverización y tamizado antes de realizar la prueba.
-- Ubicar la muestra en el espacio entre los dos cilindros de acero. Para el caso de líquidos este espacio debe ser de 1.0 mm.
-- Ajustar el equipo con la carga de 10 kg.
-- Dejarla caer desde una altura de 0.40 m.
-- Realizar seis (6) pruebas utilizando estas condiciones.
-- En caso que se presente explosión, repetir el procedimiento, empleando la carga de 5 kg desde un altura de 0.15 m.
-- Realizar una serie de seis (6) pruebas.
Sensibilidad mecánica-fricción.
-- Tomar una muestra de 10 ml de la sustancia bajo prueba. En el caso de sustancias sólidas, someterlas a secado y tamizado para la selección de partículas que pasen tamiz de malla 0.5 mm. Si la sustancia se encuentra en gránulos es necesaria su pulverización y posterior tamizado. Para sustancias pastosas se debe realizar un procedimiento de secado, pulverización y tamizado. En caso que no sea posible el secado de la sustancia, se realiza la formación de láminas de 0,5 mm de espesor, 2 mm de ancho y 10 mm de longitud.
-- Ubicar la muestra en la placa de porcelana.
-- Ubicar el cilindro de porcelana justo sobre la muestra, verificando que se encuentren en contacto.
-- Permitir el movimiento de la placa de porcelana 10 mm hacia delante y hacia atrás en un tiempo de 0.44s.
-- Realizar seis (6) pruebas utilizando una carga de 360 N, teniendo en cuenta que cada una de las partes de la superficie de la placa y el cilindro solo puede ser utilizada en una (1) ocasión, por lo que los dos extremos del cilindro servirán para dos pruebas y las dos superficies de la placa podrán ser usadas en un número igual de ensayos.
-- En caso que se presente explosión, repetir el procedimiento, empleando la carga de 120 N.
-- Realizar una serie de seis (6) pruebas.
VII. RESULTADOS
Se considera que la sustancia es explosiva por sensibilidad al choque, si en alguna de las pruebas correspondientes realizadas se presenta una explosión. En caso contrario, se cataloga la sustancia como no explosiva por sensibilidad al choque.
De la misma manera, se considera que la sustancia es explosiva por sensibilidad a la fricción si en alguna de las pruebas correspondientes realizadas se presenta una explosión. En caso contrario, se cataloga la sustancia como no explosiva por sensibilidad a la fricción.
Sensibilidad mecánica-choque.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.15. Diagrama de flujo procedimiento para determinación de sensibilidad mecánica al choque.
Sensibilidad mecánica-fricción.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.16. Diagrama de flujo procedimiento para determinación de sensibilidad mecánica a la fricción.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
Directiva de la Comunidad Europea. EC Directive 92/62/EEC. Método A14
3.6 Diagrama metodológico determinación característica de explosividad
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 3.17. Arbol de decisión para ejecución de pruebas relativas a clasificación de residuos explosivos
4. PROTOCOLOS METODOLOGICOS PARA INFLAMABILIDAD
Para la determinación de la inflamabilidad de un residuo, se cuenta con tres (3) procedimientos diferentes cuya aplicación depende del estado en el que se encuentre la sustancia a analizar (líquido, sólido o gaseoso).
Por lo tanto, para la evaluación de esta característica de peligrosidad la primera etapa consiste en la identificación del estado del residuo, continuando con la aplicación de la prueba correspondiente. La Figura 4.5 presenta un esquema global del procedimiento a emplear en el análisis de la inflamabilidad de un residuo.
A continuación se presentan los tres (3) protocolos seleccionados para la determinación de la inflamabilidad de sustancias en estado líquido, sólido o gaseoso.
4.1 Inflamabilidad de líquidos
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias en estado líquido, principalmente derivados del petróleo.
-- Permite determinar el punto de inflamabilidad (flash point) de una sustancia en un rango de temperatura de 40 a 360 ºC.
-- La determinación puede realizarse siguiendo dos (2) procedimientos: A y B. El primero es aplicable a sustancias como aceites destilados (diesel, queroseno, aceite térmico y combustible de turbina), aceites lubricante nuevos y otros líquidos del petróleo no incluidos en el alcance del procedimiento B. El procedimiento B tiene aplicabilidad en residuos de aceites usados, aceites lubricantes usados, mezclas de líquidos del petróleo y sólidos, líquidos del petróleo que tienden a formar una película en la superficie bajo las condiciones de la prueba o líquidos del petróleo de tal viscosidad cinemática que no son calentados uniformemente bajo las condiciones de agitación y calentamiento utilizadas en el procedimiento A.
II. TERMINOS Y DEFINICIONES
Punto de inflamabilidad (flash point): corresponde a la mínima temperatura corregida a presión barométrica 101.3 kPa a la cual la aplicación de una fuente de ignición causa que los vapores producidos por la muestra ardan bajo condiciones específicas de la prueba utilizada para la determinación.
III. PRINCIPIO DEL METODO
El método permite la determinación del punto de inflamación de una sustancia mediante el calentamiento progresivo de esta y la puesta en contacto con una fuente de ignición a intervalos de tiempo determinados. El procedimiento se realiza utilizando un equipo de Pensky-Martens de copa cerrada automático o manual y el rango de la determinación se encuentra entre 40 a 360 ºC.
IV. INTERFERENCIAS
No se reportan posibles interferencias en la determinación.
V. EQUIPOS Y MATERIALES
i) Equipo de Pensky-Martens de copa cerrada automático o manual;
ii) Mecanismo para medición de la temperatura: termómetro o mecanismo electrónico tal como un termómetro de resistencia o una termocupla. En el caso de termómetros, la Tabla 4.1 presenta el número indicado de instrumento.
Tabla 4.1
Termómetro a utilizar en determinación de punto de inflamabilidad
| Número de termómetro | ||
| Rango de temperatura | ASTM | IP |
| -5 -110 ºC | 9C | 15C |
| 10 – 200 ºC | 88C | 101C |
| 90 – 370 ºC | 10C | 16C |
-- Fuente de ignición: llama de gas natural, llama de gas propano o fuente de ignición eléctrica (espira caliente):
iii) Solventes de limpieza: tolueno o acetona.
VI. REACTIVOS
No se requieren.
VII. PROCEDIMIENTO
Procedimiento A-Equipo manual [5]
Primera etapa.
-- Ubicar el equipo en una superficie plana.
-- Preparar el equipo para la prueba de acuerdo a las instrucciones del fabricante respecto a calibración y revisión.
-- Limpiar y secar todas las partes y accesorios del equipo utilizando los solventes de limpieza indicados.
-- Ajustar de acuerdo a las instrucciones del fabricante el sistema de detección automático de flash point en caso que vaya a ser utilizado [6].
-- Introducir en el recipiente para muestra, mínimo 75 ml de la sustancia bajo prueba. En el caso de muestras de aceites usados, el recipiente para muestra debe llenarse hasta un 85 o 95% de su capacidad. Para otro tipo de muestras, el recipiente deberá contener muestra entre un 50 y 85% de su capacidad.
-- Introducir en la cápsula de prueba la cantidad de muestra necesaria para alcanzar la marca que se encuentra en su interior.
-- Colocar la cubierta en la cápsula y ubicar el conjunto en el equipo, asegurándose que el mecanismo de seguridad se encuentre adecuadamente ajustado.
-- Ubicar el mecanismo de medición de temperatura en el sitio apropiado.
-- Encender la llama de prueba y ajustar su diámetro a una medida de 3.2 a 4.8 mm o encender la fuente de ignición eléctrica y ajustar su intensidad de acuerdo a las instrucciones del fabricante del equipo.
-- Iniciar el calentamiento de la muestra a una velocidad tal que la temperatura se incremente entre 5 y 6 ºC cada minuto.
-- Encender el sistema de agitación a una velocidad de 90 a 120 rpm ubicando el agitador en dirección perpendicular.
-- Calentar la muestra hasta 15 ± 5 ºC [7], e iniciar la aplicación de la fuente de ignición.
-- Aplicar la fuente de ignición cada vez que la temperatura se eleve 1 ºC.
-- La aplicación de la fuente de ignición se realiza mediante la suspensión de la agitación y la operación del mecanismo en la cubierta de la cápsula de prueba que permite la obstrucción, de tal forma que la fuente de ignición se ubique en el espacio del vapor de la cápsula de prueba en 0.5 s, permanezca allí durante 1 s y regrese a su posición superior.
-- Registrar como la temperatura de inflamación aquella indicada en el mecanismo de medición de temperatura en el momento en que la aplicación de la fuente de ignición causa una llama en el interior de la cápsula de prueba y esta se extiende sobre toda la superficie de la muestra[8].
-- Cuando se utiliza una llama de gas como fuente de ignición, su aplicación puede causar un halo azul o un aumento en su tamaño, previos al flash point, por lo que al no corresponder a la temperatura de interés, estos efectos no deben ser considerados.
-- Si el flash point se detecta en la primera aplicación de la fuente de ignición, la prueba debe ser descartada y realizada nuevamente utilizando una muestra diferente y realizando la primera aplicación de la fuente de ignición a una temperatura de 23 ± 5 ºC por debajo de la temperatura a la cual se presentó la inflamación.
-- Si el flash point detectado se encuentra 28 ºC por encima de la temperatura a la cual se inicia el contacto con la fuente de ignición o a menos de 18 ºC a partir de esta temperatura, se considera que el resultado es aproximado y la prueba se debe repetir con una muestra diferente.
-- Atendiendo a que las sustancias estudiadas tienen un rango de flash point que no se conoce con anterioridad, los resultados obtenidos son aproximados y en caso de requerirse una determinación exacta, debe realizarse de manera posterior a la primera determinación el procedimiento presentado en la segunda etapa.
-- Una vez se ha registrado el flash point, se permite el enfriamiento del equipo.
-- Se remueve la cubierta de prueba y se realiza la limpieza del equipo de acuerdo a las instrucciones del fabricante.
Segunda etapa
-- Si el flash point determinado en la primera etapa se encuentra por debajo de 100 ºC, se realiza el calentamiento de la muestra y se aplica por primera vez la fuente de ignición cuando la temperatura se encuentra a 23 ± 5 ºC por debajo del punto de inflamación esperado.
-- Aplicar la fuente de ignición cada vez que la temperatura se eleve 1 ºC.
-- La aplicación de la fuente de ignición se realiza mediante la suspensión de la agitación y la operación del mecanismo en la cubierta de la cápsula de prueba que permite la obstrucción, de tal forma que la fuente de ignición se ubique en el espacio del vapor de la cápsula de prueba en 0.5 s, permanezca allí durante 1 s y regrese a su posición superior.
-- Registrar la temperatura a la cual se presenta la inflamación.
-- Si el flash point determinado en la primera etapa se encuentra por encima de 110 ºC, se aplica la fuente de ignición de la manera descrita anteriormente iniciando cuando la temperatura se encuentra a 23 ± 5 ºC por debajo del punto de inflamación esperado y a intervalos de aumento en la temperatura de 2 ºC.
Procedimiento B-Equipo manual
Corresponde a la misma secuencia y etapas del procedimiento A con la diferencia que el agitador es puesto en funcionamiento a una velocidad de 250 ± 10 rpm antes de iniciar el calentamiento de la muestra y el calentamiento posterior se realiza a una velocidad tal que la temperatura incrementa de 1 a 1.6 ºC cada minuto.
VIII. RESULTADOS
La temperatura determinada cono punto de inflamación, debe ser corregida a la presión de 101.3 kPa en caso que las condiciones de presión en la realización de la prueba no correspondan a este valor. La corrección se realiza mediante la ecuación:
Punto de inflamación corregido = C + 0.25(101.3-K) (2)
Donde:
C = punto de inflamación observado ( ºC)
K = presión atmosférica del sitio de realización de la prueba (kPa)
El reporte de los resultados debe indicar el procedimiento utilizado A o B y la temperatura de inflamación corregida a 101.3 kPa y redondeada a 0.5 ºC.
Con base en el resultado se compara con el límite establecido, que para el caso de Colombia es de 60 ºC, y se determina si la sustancia puede considerarse como inflamable.
Procedimiento A[9]
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 4.1. Diagrama de flujo procedimiento para determinación líquidos inflamables.
IX. INFORMACION ESTADISTICA
Repetibilidad.
Para el procedimiento A, esta resulta ser: 0.029X, donde X corresponde al punto de inflamación determinado expresado en ºC. Para el procedimiento B, la repetibilidad es de 2 ºC para aceites combustibles residuales y 5 ºC para otro tipo de sustancias.
Reproducibilidad.
Para el procedimiento A, esta resulta ser: 0.071X, donde X corresponde al punto de inflamación determinado expresado en ºC. Para el procedimiento B, la reproducibilidad es de 6 ºC para aceites combustibles residuales y 10 ºC para otro tipo de sustancias.
X. REFERENCIAS
Norma ASTM D:93 -02ª.
4.2 Inflamabilidad de sólidos
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias sólidas en polvo, en forma granular o pastosa.
-- Permite determinar la capacidad de una sustancia en este estado para propagar la combustión.
II. PRINCIPIO DEL METODO
En el método, la sustancia se somete a inflamación y se determina la duración y velocidad de la combustión, evaluando con base en parámetros establecidos si la sustancia puede considerarse como inflamable.
III. INTERFERENCIAS
No se reportan posibles interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
-- El método emplea un molde de sección triangular de 250 mm de longitud, con dimensiones interiores de 10 mm de altura y 20 mm de ancho (Figura 4.2). A los lados del molde de manera longitudinal se encuentran fijas láminas de metal que sobresalen 2 mm por encima del borde superior de la sección triangular.
-- Placa de material no poroso, incombustible y de baja conductibilidad térmica, que sirve como soporte para la muestra.
-- Quemador de gas con diámetro mayor o igual a 5 mm.
-- Gas combustible.
-- Pantalla de protección.
-- Campana extractora.
-- Cronómetro.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 4.2. Molde para determinación de inflamabilidad en sólidos.
V. REACTIVOS
No se requieren.
VI. PROCEDIMIENTO
Prueba preliminar.
-- Una muestra de la sustancia es dispuesta en una tira de aproximadamente 250 mm de longitud, 20 mm de ancho y 10 mm de altura sobre la placa que sirve como soporte.
-- Mediante el quemador de gas, se aplica una llama a un extremo de la tira del material a una temperatura de llama mayor a 1000 ºC.
-- Se mantiene la aplicación de la llama hasta que se presente inflamación o por un tiempo máximo de 2 minutos o 5 minutos en el caso de polvos metálicos y aleaciones de metales.
-- Se observa si la combustión se propaga a lo largo de los 200 mm iniciales de la tira durante 4 minutos o 40 minutos en el caso de polvos metálicos y aleaciones de metales
-- Si no se presenta este efecto se da por terminada la prueba.
-- En caso de observarse la propagación de la combustión dentro de los límites establecidos se sigue el procedimiento presentado en la prueba de velocidad de la combustión.
Prueba de velocidad de la combustión.
-- Si la muestra corresponde a una sustancia en polvo o gránulos, se llena el molde con la cantidad de muestra necesaria. En caso de sustancias pastosas, no se realiza la etapa de moldeo, sino que se forma sobre una superficie incombustible, un cordón de 250 mm de longitud y aproximadamente 100 mm2 de sección transversal.
-- Se deja caer el molde en tres (3) oportunidades desde una altura de 20 mm sobre una superficie sólida.
-- Se retiran las láminas laterales del molde y se coloca sobre el molde la lámina utilizada como soporte de la muestra.
-- Se invierte la posición del sistema molde-lámina y se retira el molde de manera que la muestra quede depositada sobre el soporte.
-- Se rodea el soporte con una pantalla de protección y se ubica en una campana extractora.
-- A 30 o 40 mm de los 180 mm iniciales del molde formado con la sustancia se aplica por goteo 1 ml de una solución humectante[10],[11] de manera que la sección transversal del molde se humedezca totalmente sin pérdida lateral de líquidos[12].
-- Se enciende la muestra por su extremo inicial utilizando un medio adecuado como una llama pequeña o un hilo metálico muy caliente a 1000 ºC como mínimo.
-- Cuando la muestra ha ardido hasta una distancia de 80 mm, se mide la velocidad de la combustión a lo largo de los 100 mm siguientes.
-- Se realizan seis (6) pruebas utilizando en cada caso una placa de soporte limpia y fría.
VII. RESULTADOS
Se considera que la sustancia no es inflamable si en la prueba preliminar no se inflama, ni se propaga la combustión con o sin llama durante los cuatro (4) minutos establecidos (40 para el caso de aleaciones de metales o polvos metálicos). La sustancia se cataloga como inflamable si la velocidad de combustión en los 100 mm analizados, resulta ser mayor a 2.2 mm/s. En el caso de polvos metálicos y aleaciones de metales, se considera que son inflamables cuando la reacción se propaga a toda la longitud de la muestra en un tiempo menor o igual a 10 minutos.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 4.3. Diagrama de flujo procedimiento para determinación de sólidos inflamables.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
UNECE-Manual de Pruebas y Criterios-Prueba N.1.
Directiva de la Comunidad Europea. EC Directive 92/62/EEC. Método A10.
4.3 Inflamabilidad de gasés
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias en estado gaseoso.
-- Permite determinar si un gas mezclado con aire resulta inflamable a una temperatura de 20 ºC y presión atmosférica.
II. TERMINOS Y DEFINICIONES
Rango de inflamabilidad: Rango de concentración de gas entre los límites de explosividad.
Límites de explosividad: Límites de concentración de un gas inflamable en mezclas con aire a los cuales no ocurre la propagación de una llama.
III. PRINCIPIO DEL METODO
En el método, se realizan mezclas de gas y aire a concentraciones crecientes, a temperatura de 20 ºC y presión ambiente, y se determina si son inflamables poniéndolos en contacto con una chispa eléctrica y determinando si ocurre una ignición.
IV. INTERFERENCIAS
No se reportan posibles interferencias en la determinación.
V. EQUIPOS Y MATERIALES
-- Recipiente de prueba: cilindro de vidrio con diámetro interno de 50 mm. y altura mínimo de 300 mm, acoplado con mecanismo de liberación de presión y aislado para restringir cualquier daño por explosión.
-- Electrodos de ignición, ubicados 60 mm. por encima del fondo del reactor dentro de este y separados a una distancia de 3 a 5 mm.
-- Transformador con salida de 10 a 15 kV y máxima potencia de entrada de 300 W, que permite la generación de una chispa de inducción de 0.5 s de duración.
-- Bomba proporcional.
VI. REACTIVOS
No se requieren.
VII. PROCEDIMIENTO
-- Utilizando una bomba proporcional se introduce una mezcla gas-aire con concentración de 1% v/v de gas en el cilindro de vidrio.
-- Se hace pasar una chispa a través de la mezcla.
-- Se observa si se presenta una ignición en el recipiente y si se propaga de manera independiente.
-- Se varía la concentración de gas en intervalos de 1% v/v y se repite el procedimiento.
-- Se realiza el procedimiento hasta alcanzar una concentración de 13% v/v de gas en la mezcla en caso de que no se haya producido inflamación en una prueba anterior.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 4.4. Diagrama de flujo procedimiento para determinación de gases inflamables.
VIII. RESULTADOS
Se considera que el gas es inflamable si en alguna de las pruebas realizadas se presenta una ignición en el recipiente y esta se propaga de manera independiente. En el reporte de resultados se indica la concentración de gas a la que se presentó la ignición.
Si en ninguna de las pruebas realizadas se presenta este efecto, el gas se cataloga como no inflamable en el rango de concentración estudiado.
IX. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
X. REFERENCIAS
Directiva de la Comunidad Europea. EC. Directive 92/69/EEC. Método A11.
4.4 Diagrama metodológico determinación característica de inflamabilidad
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 4.5. Arbol de decisión para ejecución de pruebas relativas a clasificación de residuos inflamables.
5. PROTOCOLOS METODOLOGICOS PARA REACTIVIDAD
Las pruebas propuestas para la determinación de la reactividad de un residuo incluyen la evaluación de la capacidad de este para experimentar combustión espontánea, calentamiento espontáneo, liberar gases inflamables al entrar en contacto con agua y exhibir propiedades comburentes.
Estas se caracterizan por ser pruebas complementarias que evalúan distintos aspectos de esta característica de peligrosidad. Por lo tanto, no existe un orden único para su ejecución, y los resultados obtenidos a través de la aplicación de cada procedimiento permiten concluir si el residuo presenta la característica específica bajo evaluación y si este puede ser considerado reactivo desde ese punto de vista.
En la Figura 5.7 se presenta un árbol de decisión que ofrece una guía para llevar a cabo los procedimientos comprendidos en la evaluación de la característica de reactividad. Esta secuencia ofrece tan solo una de las posibilidades existentes para la realización de las distintas pruebas, y es decisión de quien evalúa la característica aplicar un esquema diferente, considerando siempre que existen algunas pruebas específicas para sustancias sólidas y otras que sólo pueden ser aplicadas a sustancias en estado líquido.
A continuación se presentan los seis (6) protocolos seleccionados para la determinación de distintas características, cuya evaluación permitirá la catalogación de un residuo como reactivo.
5.1 Método de prueba para sólidos que pueden experimentar combustión espontánea
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias en estado sólido.
-- Permite determinar la capacidad de una sustancia en este estado para experimentar combustión espontánea al ser expuesta al aire.
II. PRINCIPIO DEL METODO
Una sustancia en estado sólido es puesta en distintas formas en contacto con aire durante un período de tiempo específico, con el propósito de determinar su capacidad para experimentar combustión espontánea.
III. INTERFERENCIAS
No se reportan posibles interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
Superficie incombustible.
V. REACTIVOS
No se requieren.
VI. PROCEDIMIENTO
-- Se toma una muestra de uno (1) a dos (2) mg de la sustancia en polvo.
-- Desde aproximadamente un (1) m de altura se deja caer la muestra sobre una superficie incombustible, observando si esta sufre inflamación durante el descenso.
-- Se permite a la muestra permanecer por cinco (5) minutos sobre la superficie y se registra si se presenta inflamación en este período de tiempo.
La operación se realiza seis (6) veces a menos que se observe inflamación en el transcurso de las pruebas.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 5.1. Diagrama de flujo procedimiento para determinación de sólidos que pueden experimentar combustión espontánea.
VII. RESULTADOS
Si la muestra se inflama durante el descenso o en los cinco (5) minutos siguientes a este, o mientras permanece en la superficie incombustible, se considera que la sustancia tiene la capacidad de experimentar combustión espontánea.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
UNECE-Manual de Pruebas y Criterios-Prueba N. 2.
5.2 Método de prueba para líquidos que pueden experimentar combustión espontánea
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias líquidas.
-- Permite determinar la capacidad de una sustancia en este estado para experimentar combustión espontánea.
II. PRINCIPIO DEL METODO
En el método, que consta de dos etapas, una sustancia líquida es inicialmente incorporada a un soporte inerte y expuesta al aire determinando si presenta inflamación; en caso negativo, en una segunda fase, la muestra es depositada en un trozo de papel filtro y expuesta al aire, observado su capacidad para causar la inflamación o carbonización del papel filtro.
III. INTERFERENCIAS
No se reportan posibles interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
-- Cámara de humedad y temperatura controlada.
-- Cubeta de porcelana de 100 mm. de diámetro.
-- Tierra de diatomeas o sílice gelatinosa.
-- Jeringa.
-- Papel filtro.
V. REACTIVOS
No se requieren.
VI. PROCEDIMIENTO
Primera etapa.
-- Dentro de la cubeta de porcelana se deposita la cantidad necesaria de tierra de diatomeas o sílice gelatinosa para lograr una capa de aproximadamente cinco (5) milímetros de altura.
-- Se adicionan 5 ml de la sustancia estudiada en la capa de tierra de diatomeas o sílice gelatinosa previamente formada.
-- Se observa si en un período de cinco (5) minutos se presenta inflamación de la sustancia.
-- El procedimiento se realiza seis (6) veces a menos que se observe inflamación en una prueba anterior.
-- En caso que no se presente inflamación de la muestra, se procede a realizar la prueba descrita en la segunda etapa.
Segunda etapa.
-- Mediante una jeringa se toman 0.5 ml de la sustancia a estudiar.
-- Se deposita la muestra sobre un papel filtro seco con una pequeña concavidad.
-- Se introduce el conjunto (muestra + papel filtro) en la cámara de humedad y temperatura controlada a 25 ± 2 ºC y 50 ± 5% de humedad relativa.
-- Se observa si durante un período de cinco (5) minutos, a partir que la muestra fue depositado, el papel filtro se inflama o carboniza.
La prueba se efectúa tres (3) veces a menos que se observe inflamación o carbonización del papel filtro en una prueba anterior. Se utiliza un papel filtro nuevo en cada ocasión.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 5.2 Diagrama de flujo procedimiento para determinación de líquidos que pueden experimentar combustión espontánea.
VII. RESULTADOS
Se considera que la sustancia experimenta combustión espontánea si en la primera etapa del procedimiento presenta inflamación, o si en la segunda etapa de este, se produce la inflamación o carbonización del papel filtro en el período de tiempo estipulado.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
UNECE-Manual de Pruebas y Criterios-Prueba N.3.
5.3 Método de prueba para sólidos que pueden experimentar calentamiento espontáneo
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias en estado sólido.
-- Permite determinar la capacidad de una sustancia en este estado para experimentar calentamiento espontáneo por oxidación.
II. PRINCIPIO DEL METODO
En este método, una muestra sólida es sometida a calentamiento por circulación de aire dentro de un horno a una temperatura de 140 ºC, realizando un seguimiento continuo a su temperatura con el fin de determinar su capacidad para experimentar calentamiento espontáneo.
III. INTERFERENCIAS
No se reportan interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
-- Horno de aire caliente circulante, con volumen mayor a 9 L, provisto de dispositivos de control para mantener la temperatura interna a 140 ± 2 ºC
-- Registrador electrónico de datos (datalogger) para temperatura.
-- Dos portamuestras metálicos de 25 x 100 mm de lado, elaborados en acero inoxidable con malla de 0.05 mm y abiertos en la parte superior.
-- Receptáculo cúbico de tela de acero inoxidable malla 0.6 mm, con un tamaño un poco mayor al del portamuestras, de tal forma que permita la disposición de este en su interior.
-- Receptáculo de tela de acero inoxidable con malla 0.595 mm, con las siguientes dimensiones: 150 x 150 x 250 mm.
-- Dos termopares de cromel–alumel de 0.3 mm de diámetro.
V. REACTIVOS
No se requieren.
VI. PROCEDIMIENTO
-- La sustancia en polvo o en forma granular (estado original) es introducida en el portamuestras hasta alcanzar el borde de este, golpeando varias veces para comprimir la muestra (si la muestra se lograr comprimir se debe adicionar cantidad de muestra adicional hasta alcanzar nuevamente el borde del portamuestras).
-- El portamuestras es introducido en el receptáculo de malla 0.6 mm.
-- El receptáculo es suspendido en el centro del horno.
-- Se introduce en el centro de la muestra uno de los termopares disponibles.
-- Se ubica el termopar adicional en un punto entre el portamuestras y la pared del horno.
-- Se ajusta la temperatura del horno a 140 ºC y se mantiene en este nivel durante 24 h.
-- A intervalos de 1 minuto y durante 24 h se registran la temperatura de la muestra y el horno, utilizando el datalogger.
VII. RESULTADOS
Si en el transcurso de la prueba, se produce inflamación espontánea de la muestra, o en algún momento durante las 24 h de medición, la temperatura de esta excede en 60 ºC la temperatura del horno, se considera que la sustancia es susceptible de experimentar calentamiento espontáneo.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
UNECE-Manual de Pruebas y Criterios-Prueba N.4.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 5.3. Diagrama de flujo procedimiento para determinación de sólidos que pueden experimentar calentamiento espontáneo.
5.4 Método de prueba para sustancias que en contacto con agua desprenden gases inflamables
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable a sustancias sólidas y líquidas incluyendo sustancias que tienen la capacidad de experimentar combustión espontánea[13]
-- Permite determinar la capacidad de una sustancia para desprender gases inflamables al entrar en contacto con agua.
II. PRINCIPIO DEL METODO
En este método, una sustancia en estado sólido o líquido es puesta en contacto con agua en diferentes formas, de tal manera que sea posible establecer si en alguna de estas se presenta el desprendimiento de gases que se inflaman espontáneamente o si los gases inflamables se desprenden a una velocidad mayor a 1 L/kg sustancia.h.
III. INTERFERENCIAS
No se reportan interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
-- Cubeta.
-- Papel filtro.
-- Cápsula de evaporación de 100 mm de diámetro.
-- Cronómetro.
-- Gotero.
-- Embudo.
-- Erlenmeyer con desprendimiento lateral.
-- Tamiz de abertura 0.5 mm.
-- Caudalímetro para gases o rotámetro
V. REACTIVOS
-- Agua destilada.
VI. PROCEDIMIENTO
Primera etapa.
-- Se introduce agua destilada en una cubeta.
-- Se deposita en la cubeta una cantidad de muestra equivalente a aproximadamente 2 mm de diámetro.
-- Se observa si por el contacto con el agua se produce algún desprendimiento de gas y si este se inflama espontáneamente. Si no se presenta este comportamiento se continúa con el procedimiento descrito en la segunda etapa.
Segunda etapa.
-- En la cápsula de evaporación se deposita agua destilada.
-- La misma cantidad de muestra utilizada en la primera etapa es depositada en un papel filtro extendido.
-- El papel filtro con la muestra es dispuesto en la cápsula de evaporación, de forma que quede extendido y se mantenga sobre la superficie del agua en una posición fija.
-- Se observa si por el contacto con el agua se produce algún desprendimiento de gas y si este se inflama espontáneamente. Si no se presenta este comportamiento y la sustancia bajo prueba se encuentra en estado sólido, se continúa con el procedimiento descrito en la tercera etapa. En caso contrario se sigue el procedimiento descrito en la cuarta etapa.
Tercera etapa.
-- Se toma una cantidad de muestra suficiente para formar un montículo de aproximadamente 20 mm de altura y 30 mm de diámetro.
-- En la parte superior del montículo se abre un agujero pequeño.
-- En el agujero se vierten cinco (5) gotas de agua.
-- Se observa si por el contacto con el agua se produce algún desprendimiento de gas y si este se inflama espontáneamente.
-- Si no se registra desprendimiento de gas e inflamación de este, se continúa con el procedimiento correspondiente a la cuarta etapa.
Cuarta etapa.
Si la sustancia no presenta reacción violenta con el agua según los resultados obtenidos en las etapas anteriores, debe seguirse el procedimiento descrito a continuación:
-- Si se trata de una sustancia en estado sólido, se examina el porcentaje de partículas de tamaño menor a 500 ím de diámetro que contiene la sustancia (porcentaje en peso).
-- Si el porcentaje es mayor a 1%, la muestra debe pulverizarse antes de iniciar la cuarta etapa del procedimiento. En caso contrario se utiliza en su forma original.
-- En un erlenmeyer con desprendimiento lateral, se deposita una cantidad de muestra menor a 25 g, registrando el peso exacto de esta.
-- En la boca del erlenmeyer se ubica un embudo de llave.
-- Se deposita agua en el embudo.
-- Se abre la llave del embudo permitiendo el contacto del agua con la muestra.
-- Se mide el volumen del gas desprendido empleando un procedimiento adecuado y se toma el tiempo que transcurre hasta que dejan de desprenderse gases.
-- La prueba se realiza tres (3) veces.
Para verificar si los gases desprendidos son inflamables, debe seguirse el procedimiento descrito para determinación de gases inflamables.
VII. RESULTADOS
Si la muestra presenta desprendimiento de gases que se inflaman espontáneamente en alguna etapa de la prueba o si la velocidad de desprendimiento de gas (V) calculada como:
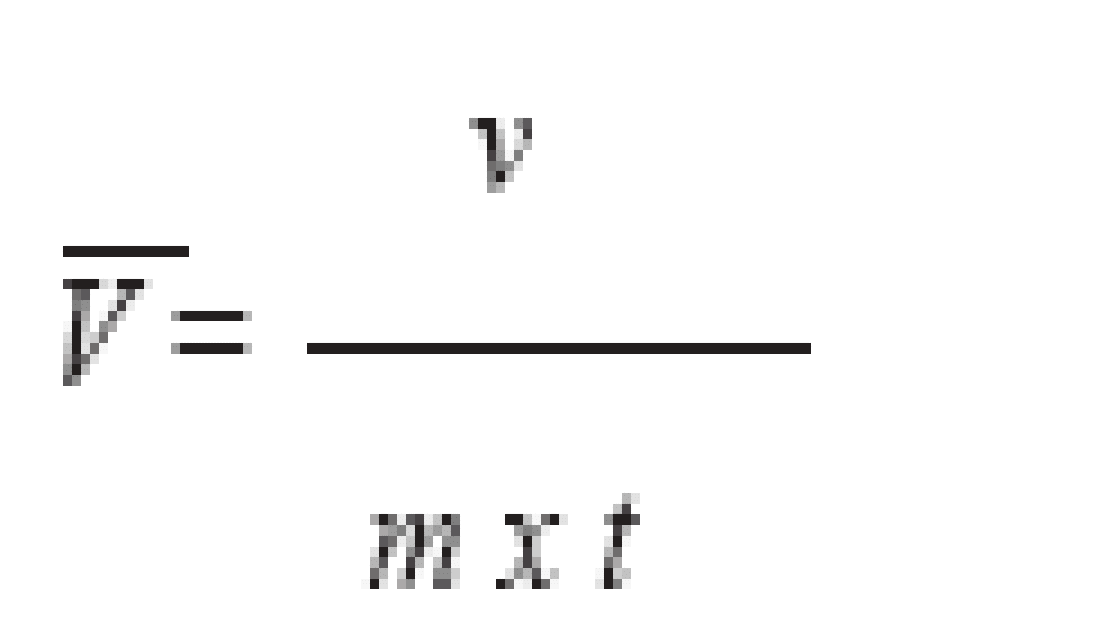
Donde:
| V: | velocidad de liberación de gas (L/kg.h) |
v: | volumen de gas desprendido durante el período de tiempo (L) |
m: | peso de la muestra introducida en el erlenmeyer (kg) |
| t: | tiempo de medición (h). |
Si la velocidad de desprendimiento es mayor a 1 l/kg sustancia.h, se considera que la sustancia desprende gases inflamables en contacto con agua.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 5.4. Diagrama de flujo procedimiento para determinación de residuos que por contacto con agua desprenden gases inflamables.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
UNECE-Manual de Pruebas y Criterios-Prueba N.5.
5.5 Método de prueba para sustancias sólidas comburentes[14]
I. ALCANCE Y APLICABILIDAD
-- El método es aplicable exclusivamente a sustancias sólidas.
-- Permite determinar si una sustancia en este estado tiene la capacidad para aumentar la velocidad o intensidad de combustión de una sustancia combustible con la que forme una mezcla homogénea.
II. PRINCIPIO DEL METODO
El método permite determinar si una sustancia sólida se comporta como comburente al encontrarse mezclada homogéneamente con un material combustible. La determinación se realiza mediante la formación de mezclas de la sustancia con celulosa en distintas proporciones y la comparación de las características de combustión con las exhibidas por una mezcla patrón de bromato de potasio y celulosa. Si la mezcla problema muestra un tiempo de combustión igual o menor al de la mezcla patrón, o se inflama o quema durante la prueba, se reporta la sustancia como comburente.
III. INTERFERENCIAS
No se reportan interferencias en la determinación.
IV. EQUIPOS Y MATERIALES
-- Celulosa fibrosa con longitud de fibra de 50 a 250 µm y diámetro medio de 25 µm.
-- Fuente de inflamación constituida por un hilo de metal inerte (níquel, cromo) conectado a una fuente de energía eléctrica. La longitud del hilo debe ser de 30 ± 1 cm, diámetro de 0.6 ± 0.05 mm. y resistencia eléctrica de 6.0 ± 0.5 O/m. El hilo debe tener la forma que se presenta en la Figura 5.5 y la potencia disipada por este debe ser de 150 ± 7 W.
-- Embudo de vidrio con ángulo de 60o y diámetro de 70 mm, sellado en su abertura inferior.
-- Placa cuadrada de 150 mm de lado y 6 mm de espesor, elaborada en un material de baja conductividad térmica (0.23 W/m.K).
-- Campana de extracción.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 5.5. Configuración experimental para prueba de determinación de sólidos comburentes.
V. REACTIVOS
-- Bromato de potasio grado reactivo (polvo que pase tamiz de abertura 0.30 mm)
VI. PROCEDIMIENTO
-- Secar aproximadamente 100 g de bromato de potasio a 65 ºC hasta peso constante (mínimo 12 horas).
-- Conservar en desecador el bromato de potasio hasta el momento de uso.
-- Secar aproximadamente 300 g de celulosa fibrosa a 105 ºC hasta que el porcentaje de humedad en el material sea inferior a 0.5% en base seca.
-- Conservar en desecador hasta el momento de uso.
-- Con el fin de dar a las muestras forma de cono truncado con 70 mm de diámetro en la base, utilizar el embudo sellado en su abertura inferior para formar 30 ± 0.1 g de mezcla de referencia: bromato de potasio/celulosa en relación 3:7 en peso. El moldeo de las muestras debe realizarse por medios mecánicos, sin aplicar excesiva presión y debe utilizarse en el menor tiempo posible.
-- Disponer la placa de baja conductividad con el hilo de ignición en su superficie, en una campana de extracción.
-- Ubicar el cono correspondiente a la mezcla de bromato de potasio/celulosa: 3/7 sobre el hilo de ignición, tal como se indica en la Figura 5.5.
-- Conectar el hilo de ignición a la fuente de corriente eléctrica y mantener la corriente hasta que la muestra se queme o inflame, o durante tres (3) minutos si no se produce alguna de estas reacciones.
-- Registrar el tiempo de combustión, tomando como momento inicial aquel en el que empieza el paso de corriente por el sistema y como punto final, aquel en que se presenta la reacción principal (inflamación o quema de la muestra), sin considerar las reacciones intermitentes tales como chispas que pueden presentarse de manera posterior a la reacción principal.
-- Si el hilo de ignición se rompe en el transcurso de la prueba, esta debe repetirse.
-- El procedimiento se realiza cinco (5) veces utilizando una muestra nueva en cada oportunidad.
-- Una vez se han realizado las pruebas correspondientes a la mezcla de referencia, se efectúa nuevamente el mismo procedimiento utilizando la sustancia problema en mezcla con celulosa en proporciones en peso de 1:1 y 4:1. Estas mezclas se preparan de manera independiente y deben utilizarse en el menor tiempo posible.
-- Para cada una de las mezclas se realiza el procedimiento en cinco (5) oportunidades.
VII. RESULTADOS
Si la sustancia en mezclas con celulosa, en proporciones en peso de 1:1 o 4:1 no se inflama o quema durante tres (3) minutos, o el tiempo promedio de combustión (calculado como el promedio de los tiempos de combustión obtenidos en las cinco pruebas realizadas) es mayor al registrado por la mezcla de referencia, se considera que la sustancia no es comburente, es decir, que en mezcla homogénea con sustancias combustibles no tiene la capacidad de aumentar la velocidad o intensidad de la combustión. En caso que el tiempo promedio de combustión de las mezclas de sustancia problema/celulosa en proporciones en peso de 1:1 o 4:1 sea igual o menor al registrado para la mezcla de referencia, o la sustancia se queme o inflame durante los tres minutos de la prueba, el reporte debe indicar que la sustancia es comburente.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 5.6. Diagrama de flujo procedimiento para determinación de sólidos comburentes.
VIII. INFORMACION ESTADISTICA
No se presenta información estadística sobre los resultados obtenidos mediante la aplicación del procedimiento presentado.
IX. REFERENCIAS
UNECE-Manual de Pruebas y Criterios-Prueba O.1.
5.6 Diagrama metodológico determinación característica de reactividad
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 5.7. Arbol de decisión para ejecución de pruebas relativas a clasificación de residuos reactivos.2
6. ROTOCOLOS METODOLOGICOS PARA TOXICIDAD
Para la evaluación de la toxicidad de un residuo, se han considerado seis pruebas diferentes: TCLP, SPLP, bioluminiscencia bacterial, quimioluminiscencia bacterial, toxicidad aguda para Daphnia y ensayo de inhibición de algas.
Estos ensayos, como se ha mencionado anteriormente estudian dos enfoques diferentes de la toxicidad de un residuo, aquel referido a la salud humana bien sea en forma directa o indirecta, que incluye los ensayos de TCLP, SPLP, bioluminiscencia bacterial y quimioluminiscencia bacterial, y aquel que involucra la toxicidad hacia el medio ambiente, específicamente la toxicidad acuática, que se basa en los procedimientos de toxicidad aguda para Daphnia y ensayo de inhibición de algas.
El orden de ejecución de estos procedimientos se presenta en la Figura 6.8. En esta, se advierte que la primera prueba a ejecutar corresponde bien al ensayo de TCLP o de SPLP, dependiendo de la forma en la cual se realizará la disposición del residuo. A continuación, y como complemento a la evaluación de toxicidad a la salud humana, se procede a realizar los ensayos de bioluminiscencia bacterial y quimioluminiscencia bacterial, Posteriormente, se lleva a cabo el ensayo de toxicidad aguda para Daphnia y ensayo de inhibición de algas, con los cuales queda evaluada la toxicidad del residuo hacia el medio ambiente, específicamente su toxicidad acuática.
A continuación se presentan los protocolos seleccionados para la determinación de distintas características, cuya evaluación permitirá la catalogación de un residuo como tóxico.
6.1 Procedimiento de lixiviación para la característica de toxicidad-TCLP
I. ALCANCE Y APLICABILIDAD
-- Este método presenta el procedimiento para la obtención de un extracto TCLP. No presenta los métodos de ensayo de laboratorio para cada compuesto individual.
-- Este método está diseñado para determinar la movilidad de los analitos orgánicos e inorgánicos presentes en un desecho líquido, sólido o de múltiples fases.
-- Si el análisis total de un desecho demuestra que los analitos individuales no están presentes en el desecho, o que están presentes pero a tan bajas concentraciones que no es posible que los niveles regulatorios apropiados sean excedidos, no es necesario realizar este procedimiento de extracción.
-- Si un análisis de una de las fracciones líquidas del extracto TCLP indica que un compuesto regulado está presente en tan alta concentración, que aún después de considerar la dilución de la otra fracción del extracto, la concentración sería mayor al nivel regulatorio de aquel compuesto, el desecho se clasifica como peligroso y no es necesario analizar la fracción remante del extracto.
-- Si un análisis de extracto obtenido usando un extractor de botella muestra que la concentración de cualquier analito volátil regulado excede el nivel regulatorio para ese compuesto, el desecho se clasifica como peligroso y no es necesario la extracción usando el ZHE. Sin embargo el extracto de un extractor de botella no puede ser usado para demostrar que la concentración de compuestos volátiles está por debajo del nivel regulatorio.
II. PRINCIPIO DEL METODO
-- Desechos líquidos (aquellos que contienen menos de 0,5% de sólidos secos)
El Extracto TCLP es la fase líquida obtenida después de la filtración del desecho a través de un papel fibra de vidrio de 0,6 a 0,8 µm.
-- Desechos conteniendo mayor o igual cantidad de 0,5% de sólidos
El desecho es filtrado y el líquido filtrado corresponde a extracto TCLP fase líquida y se almacena. La fase sólida del desecho se reduce de tamaño (si es requerido), y se realiza un proceso de extracción al ponerla en contacto con un fluido de extracción (este es función de la alcalinidad de la fase sólida del desecho) igual a 20 veces su peso seco durante 18 ± 2 horas a 30 ± 2 rpm. Después del período de extracción la fase sólida con el fluido son filtradas separando el extracto líquido. Si la fase líquida y el extracto líquido son compatibles estas se mezclan y se analizan juntas. Si son incompatibles los líquidos son analizados por separado y los resultados son combinados matemáticamente para obtener una concentración promedia ponderada por volumen.
-- Desechos 100% sólidos
El desecho se reduce de tamaño (si es requerido), y se realiza un proceso de extracción al ponerla en contacto con un fluido de extracción (este es función de la alcalinidad de la fase sólida del desecho) igual a 20 veces su peso seco durante 18 ± 2 horas a 30 ± 2 rpm. Después del período de extracción la fase sólida con el fluido son filtradas separando el extracto líquido. El extracto líquido es el Extracto TCLP y se analiza.
III. INTERFERENCIAS
No se reportan interferencias sobre el método de extracción. Interferencias en el análisis cuantitativo de cada uno de los diferentes analitos deben ser asociadas a los métodos de análisis correspondientes.
IV. EQUIPOS Y MATERIALES
-- Aparato de agitación: este debe ser capaz de rotar los recipientes de extracción en rotación completa a 30 ± 2 rpm (Ver Anexo 2).
-- Recipientes de extracción:
– Recipiente de extracción de cero espacio de cabeza – (zero-headspace extraction vessel) ZHE (Ver Anexo 3).
Este se usa solo cuando se desea evaluar la movilidad de analitos volátiles en el desecho. El ZHE permite la separación líquido/sólido dentro del equipo, excluyendo el espacio de cabeza. Este tipo de recipiente permite la separación líquido/sólido inicial, extracción, y filtración del extracto final sin abrir el recipiente. Este recipiente debe tener un volumen interno de 500 a 600 ml, y estar equipado para acomodar un filtro de 90 a 110 mm. El recipiente contiene empaques de VITON los cuales deben ser reemplazados frecuentemente. En el Anexo 3 se presentan los recipientes ZHE conocidos por la EPA.
Para que el ZHE sea aceptado, el pistón interno debe ser capaz de moverse aproximadamente a 15 PSI o menos. Si se requiere más presión, los empaques deben ser reemplazados, si con esto no se resuelve el problema, el ZHE no se acepta para el análisis de TCLP y el fabricante debe ser contactado.
Después de cada extracción debe verificarse que no presente fugas, si el equipo tiene un medidor de presión, se debe presurizar hasta 50 PSI, dejar quieto por 1 hora, sumergir en agua y verificar la presencia de burbujas de aire escapando de cualquier accesorio. Si se pierde la presión verificar todos los accesorios e inspeccionar y reemplazar los empaques, si es necesario. Reensayar el mecanismo. Si los problemas de fugas no se pueden resolver, se debe contactar al fabricante.
– Recipientes de extracción de botella
Si se va avaluar compuestos no-volátiles en el desecho, se requiere un contenedor con suficiente capacidad para retener la muestra y el fluido de extracción. En este se permite espacio de cabeza.
Las botellas de extracción pueden ser de diferentes materiales, dependiendo de los analitos a ser analizados y la naturaleza del desecho. Se recomienda usar botellas de vidrio, botellas plásticas de politetrafluoroetileno (PTFE) cuando se va a analizar tanto compuestos inorgánicos y orgánicos. Si solo se van a analizar metales se pueden usar recipientes de polietileno de alta densidad (HDPE), polipropileno (PP) o cloruro de polivinilo (PVC), y se recomiendan botellas de vidrio de borosilicato.
-- Equipos de filtración (se recomienda que todas las filtraciones sean realizadas bajo campana de extracción).
– Recipiente de extracción de cero espacio de cabeza ZHE
Cuando se vayan a analizar compuestos volátiles se debe usar este equipo para la filtración. El equipo debe ser capaz de soportar y mantener en su lugar el papel de filtro de fibra de vidrio y ser capaz de resistir la presión necesaria para lograr la separación (50 PSI).
Cuando se sospeche que el papel filtro de fibra de vidrio se ha roto, un papel filtro de fibra de vidrio en línea puede ser usado para filtrar el material dentro del ZHE.
– Bomba de vacío
Puede solo ser usada para filtración de desechos con un bajo contenido de sólidos (<10%) y desechos altamente granulares, cuando se van a analizar compuestos no-volátiles. El equipo debe tener un volumen interno de 300 ml y permitir acomodar un papel filtro como mínimo de 47 mm (se recomiendan los retenedores de capacidad interna de 1,5 L o mayor y equipados para acomodar filtros de diámetro de 142 mm). En el Anexo 4 se presentan algunos equipos conocidos de la EPA. Los materiales de los equipos pueden ser vidrio, PTFE, o acero inoxidable; si solo se van a evaluar metales se acepta HDPE, PP o PVC y se recomienda vidrio borosilicato.
– Equipos de filtración de presión positiva (capaces de alcanzar hasta 50 PSI)
Se usan para desechos con alto contenido de sólidos (>10%). El equipo debe tener un volumen interno de 300 ml y permitir acomodar un papel filtro como mínimo de 47 mm (se recomiendan los retenedores de capacidad interna de 1,5 L o mayor y equipados para acomodar filtros de diámetro de 142 mm). En el Anexo 4 se presentan algunos equipos conocidos de la EPA. Los materiales de los equipos pueden ser vidrio, PTFE, o acero inoxidable si solo se va avaluar metales se acepta HDPE, PP o PVC, y se recomienda vidrio borosilicato.
-- Papel filtro
Fibra de vidrio de borosilicato con un tamaño de poro efectivo entre 0,6 a 0,8 µm. Cuando se evalúa la movilidad de metales, los filtros deben ser enjuagados con ácido (enjuagar con ácido nítrico 1N seguido por tres enjuagues consecutivos de agua destilada desionizada, se recomienda como mínimo 1L) antes de usarse. Los filtros conocidos por la EPA se presentan en el Anexo 4.
-- pH-metro
Debe garantizar una exactitud de ± 0,05 unidades a 25 ºC
-- Mecanismo de recolección del Extracto ZHE
Bolsas Tedlar®, o jeringas sello de gas de vidrio, acero inoxidable o PTFE. Para la selección de este tener en cuenta las siguientes especificaciones:
– Si el desecho contiene una fase líquida acuosa o si el desecho no contiene una cantidad significativa de líquido no acuoso (< 1% del desecho total), se debe usar bolsa Tedlar® o jeringa de 600 ml para recolectar y combinar el líquido inicial y el extracto del sólido.
– Si el desecho contiene una cantidad significativa de líquido no acuoso en la fase líquida inicial (> 1% del desecho total), se puede usar jeringa o bolsa Tedlar® tanto para la separación líquido-sólido inicial y el extracto de la filtración final. Sin embargo se debe usar una o la otra y no ambas.
– Si el desecho no contiene fase líquida inicial (es 100% sólido) o no tiene una fase sólida significativa (es 100% líquido, o <0,5% de sólidos secos) se puede usar bolsa Tedlar® o jeringa. Si se usa jeringa descartar los primeros 5 ml del líquido obtenidos del ZHE. Las demás alícuotas son usadas para el análisis.
-- Mecanismo de transferencia del fluido de extracción al ZHE
Cualquier dispositivo capaz de transferir el fluido de extracción dentro del ZHE sin cambiar la naturaleza de este (por ejemplo una bomba peristáltica o de desplazamiento positivo, una jeringa con sello, unidad de filtración de presión u otro mecanismo).
-- Balanza de laboratorio
Que garantice una exactitud de ± 0,01 g.
-- Vaso de precipitado o erlenmeyer de vidrio de 500 ml
-- Vidrio de reloj, de un diámetro apropiado para cubrir el vaso o erlenmeyer
-- Agitador magnético
-- Estufa o plancha de calentamiento.
-- Centrífuga (opcional como ayuda de filtración)
V. REACTIVOS
Todos los reactivos que se deben utilizan son grado reactivo a menos que se mencione explícitamente otro grado.
-- Agua grado reactivo
Para analitos no-volátiles agua Tipo II ASTM o equivalente. Para extracciones de compuestos volátiles se recomienda que el agua grado reactivo sea generada al pasar agua potable a través de un filtro de cama de carbón activado conteniendo alrededor de 500 gramos de este; o con un sistema de purificación (Millipore Super-Q o equivalente); o por ebullición de agua por 15 minutos, después y mientras se mantiene a 90 ± 5 ºC burbujear un gas inerte libre de contaminantes (p.ej. nitrógeno) por 1 hora, mientras está caliente transferir a una botella de boca angosta sin espacio de cabeza y sellar con septa de teflón y tapar.
-- Acido Clorhídrico 1N
-- Acido Nítrico 1N
-- Hidróxido de sodio 1N
-- Fluidos de extracción
– Fluido de extracción No 1.
Diluir 5,7 ml de ácido acético glacial concentrado (17,5 N) en 500 ml de agua grado reactivo y adicionar 64,3 ml de NaOH 1N y aforar a un litro. El pH debe ser 4,93 ± 0,05
– Fluido de extracción No 2
Diluir 5,7 ml de ácido acético glacial concentrado con agua grado reactivo hasta un volumen de un litro. El pH debe ser de 2,88 ± 0,05
VI. PROCEDIMIENTO
VI-i. Evaluaciones preliminares.
Se requiere una alícuota del desecho mínimo de 100 gramos. Esta no se utilizará para obtener el extracto TCLP.
VI-i.1 Determinación preliminar del porcentaje de sólidos.
Este es definido como la fracción de la muestra de la cual no se puede forzar a salir más líquido por la presión aplicada, siguiendo el procedimiento descrito a continuación.
-- Para desechos 100% sólidos ir al punto 3 ensayo preliminar de reducción de tamaño.
-- Si la muestra es líquida o de múltiples fases, se requiere hacer una separación líquido/sólido.
-- Prepesar el filtro y el envase que recibirán el líquido filtrado.
-- Ensamblar el equipo de filtración y el filtro. Colocar el filtro sobre el soporte y asegurar.
-- Pesar una alícuota del desecho y registrar el peso (mínimo 100 g).
-- Permitir que la fase sólida se asiente. Si el desecho se sedimenta muy lentamente este puede ser centrifugado antes de la filtración. La centrifugación debe ser usada únicamente como una ayuda de filtración; si se usa, el líquido debe ser decantado y filtrado, y después filtrar la porción sólida a través del mismo sistema de filtración.
-- Cuantitativamente transferir la muestra del desecho al retenedor del filtro (fase líquida y sólida). Dispersar la muestra del desecho uniformemente sobre la superficie del filtro.
-- Gradualmente aplicar vacío o presión de 1 a 10 PSI, hasta que aire o gas presurizado se mueva a través del filtro, si esto no se alcanza a los 10 PSI y/o si no se observa el paso de líquido en un período de 2 minutos, lentamente incrementar la presión en incrementos de 10 PSI hasta un máximo de 50 PSI. Cuando el gas presurizado comienza a moverse a través del filtro, o cuando el flujo del líquido ha cesado a 50 PSI se detiene la filtración.
-- El material retenido en el filtro es definido como la fase sólida del desecho y el filtrado como la fase líquida[15]
-- Determinar el peso de la fase líquida restando el peso del erlenmeyer vacío, del peso del erlenmeyer lleno con el filtrado después de la filtración.
-- Determinar el peso de la fase sólida de la muestra restando el peso de la fase líquida del peso total de la muestra, registrado anteriormente.
-- Registrar el peso de las fases líquida y sólida. Calcular el % de sólidos (%S) como sigue:
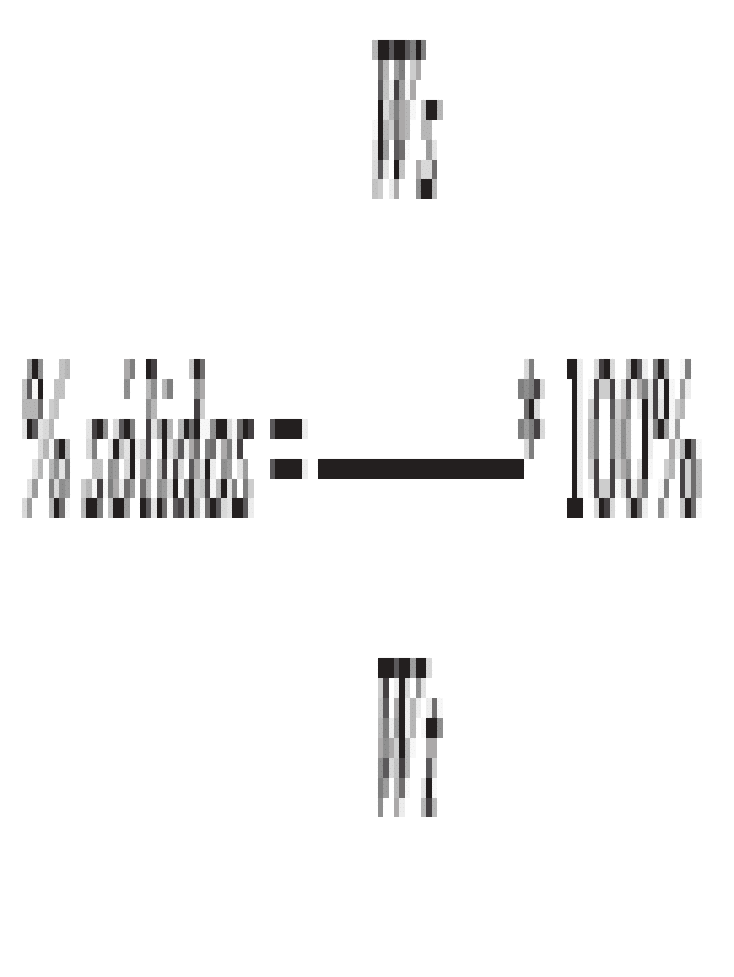
Donde:
Ws | Peso sólido |
Wt | Peso total de la muestra. |
-- Si el porcentaje de sólidos es menor de 0,5%, proceder al ensayo para no-volátiles y volátiles donde el filtrado será el extracto TCLP.
-- En el caso en el que el porcentaje de sólidos sea mayor a 0,5% y se observe que una pequeña cantidad del filtrado esta contiene agua en el filtro seguir con la determinación del porcentaje de sólidos secos, si no se observa esa pequeña cantidad de humedad pasar a la determinación de reducción de tamaño.
VI-i.2 Determinación del porcentaje de sólidos secos
-- Remover la fase sólida y el filtro del aparato de filtración.
-- Secar la fase sólida y el filtro a 100 ± 20 ºC, hasta peso constante (hasta que dos mediciones sucesivas produzcan el mismo valor dentro de ± 1%), registrar el peso final.
-- Calcular el porcentaje de sólidos secos con la siguiente ecuación.

Donde:
Ws | Peso del filtro y del desecho seco |
Wt | Peso inicial del desecho. |
-- Si el porcentaje de sólidos secos es menor de 0,5%, entonces proceder al ensayo para no-volátiles y volátiles.
-- Si el porcentaje de sólidos secos es mayor a 0,5% y si se va a realizar extracción para compuestos no-volátiles iniciar el ensayo preliminar 1 con una porción fresca del desecho para determinar si se requiere reducción de tamaño de partícula. Si solo se va a realizar extracción para volátiles seguir con el procedimiento para determinación del fluido de extracción.
VI-i.3 Determinación si el desecho requiere reducción de tamaño de partículas
Para desechos con un porcentaje de sólidos = 0.5 % o un desecho 100 % sólido se debe evaluar si este requiere reducción de tamaño de partículas. Este se requiere cuando los sólidos tienen un área superficial por gramo de material igual o mayor de 3,1 cm2, o si los sólidos presentan un tamaño mayor de 0,95 cm en su menor dimensión. Se puede realizar la evaluación a la vista, o en el caso de tener dudas pasar la fase sólida a través de un tamiz estándar de 9,5 mm (0,375 pulgadas), si quedan partículas retenidas, se requiere que a la fase sólida de la muestra en el ensayo se le haga reducción de tamaño de partícula machacándola, cortándola, moliéndola hasta llegar al área superficial o tamaño requerido
VI-i.4 Determinación del fluido apropiado para la extracción
Este numeral aplica para muestras con porcentaje de sólidos = 0,5 % y si se va a realizar extracción para constituyentes no-volátiles. Para muestras que requieran extracción de compuestos volátiles se usa solo el fluido de extracción No 1 (Ver Reactivos), y se sigue con el proceso de ensayo para compuestos volátiles.
-- Pesar una pequeña submuestra de la fase sólida del desecho, realizar reducción de tamaño de partícula (si es requerido) y tomar 5 gramos en un beaker o erlenmeyer de 500 ml.
-- Agregar 96,5 ml de agua grado reactivo al vaso, cubrir con un vidrio de reloj y agitar vigorosamente por cinco (5) minutos utilizando un agitador magnético.
-- Medir y registrar el pH. Si el pH < 5, el fluido de extracción a utilizar con la muestra es el No 1
-- Si el pH > 5, agregar 3,5 ml de HCl 1N, mezclar brevemente, cubrir con vidrio de reloj y calentar hasta 50 ºC, mantener la temperatura por 10 minutos.
-- Dejar enfriar la solución a temperatura ambiente y registrar el pH. Si el pH es < 5 utilizar el fluido de extracción No 1, si el pH es > 5 utilizar el fluido de extracción No 2
VI-ii. Ensayo
VI-ii.1 Procedimiento para extracción de compuestos no-volátiles
Se requiere un tamaño de muestra como mínimo de 100 gramos (de fase sólida y líquida). En algunos casos, puede ser apropiado una gran cantidad de muestra, dependiendo del contenido de sólidos, de si la fase líquida inicial es miscible con el extracto de la fase sólida, y de la cantidad de analitos de interés (inorgánicos, compuestos orgánicos semivolátiles, pesticidas y herbicidas). En estos casos se requiere de bastantes sólidos para generar suficiente volumen de extracto TCLP; si la cantidad de extracto generado en una extracción no es suficiente, se requerirá más de una extracción y los extractos deben ser combinados y de este obtener alícuotas para los diferentes análisis.
Si el desecho es 100% sólido pesar una submuestra del desecho, realizar reducción de tamaño si se requiere, y luego pasar a la etapa de extracción con el fluido seleccionado.
VI-ii.1.1 Filtración.
Si las muestras son líquidas o de múltiples fases, se requiere separación líquido sólido.
-- Prepesar recipiente que recibirá el filtrado.
-- Ensamblar el equipo de filtración. Colocar el filtro sobre el soporte y asegurar. Enjuagar el filtro con ácido si se va a evaluar la movilidad de metales[16]
-- Pesar una alícuota del desecho y registrar el peso (mínimo 100 g).
-- Permitir que la fase sólida se asiente. Si el desecho se sedimenta muy lentamente estos pueden ser centrifugados antes de la filtración. La centrifugación puede ser usada solo como una ayuda de filtración, si esta se usa el líquido debe ser decantado y filtrado y luego la porción sólida a través del mismo sistema de filtración.
-- Cuantitativamente transferir la muestra del desecho al retenedor del filtro (fase líquida y sólida). Dispersar la muestra del desecho uniformemente sobre la superficie del filtro[17].
-- Para muestras con un porcentaje de sólidos < 10% se puede usar bomba de vacío, para las que presentan un mayor contenido de sólidos se debe usar una bomba de presión positiva. Gradualmente aplicar vacío o presión de 1 a 10 PSI, hasta que aire o gas presurizado se mueva a través del filtro. Si esto no se alcanza a los 10 PSI y si no se observa el paso de líquido en un período de 2 minutos, lentamente incrementar la presión en incrementos de 10 PSI hasta un máximo de 50 PSI. Cuando el gas presurizado comienza a moverse a través del filtro, o cuando el flujo del líquido ha cesado a 50 PSI se detiene la filtración[18].
-- El material retenido en el filtro es definido como la fase sólida del desecho y el filtrado como la fase líquida[19].
-- Determinar el peso de la fase líquida restando el peso del erlenmeyer vacío, del peso del erlenmeyer lleno con el filtrado después de la filtración.
-- El filtrado puede ser analizado o almacenado a 4 ºC hasta el momento del análisis.
-- Si el desecho contiene < 0,5% de sólidos secos, el filtrado se define como el “Extracto TCLP”. Si el desecho contiene > 0,5% de sólidos secos, se debe almacenar o analizar la fase líquida y la fase sólida se le debe realizar el proceso de extracción después del de reducción de tamaño (si lo requiere acorde a las pruebas preliminares).
VI-ii.1.2 Reducción de tamaño fase sólida
Este proceso se aplica si la fase sólida requiere reducción de tamaño con base en las pruebas preliminares. Toda la fase sólida debe ser preparada machacando, cortando, moliendo hasta lograr las condiciones de tamaño de partícula.
VI-ii.1.3 Extracción de la fase sólida
Para desechos con > 0,5% de sólidos secos, y que no requieran reducción de tamaño de sólidos:
-- Transferir cuantitativamente la fase sólida al contenedor de extracción junto con el filtro usado en la separación líquido sólido.
Para desechos con > 0.5% de sólidos secos que requieren reducción de tamaño de partículas:
-- Transferir cuantitativamente la fase sólida después de la reducción de tamaño al contenedor de extracción junto con el filtro usado en la separación líquido sólido.
-- Determinar la cantidad del fluido de extracción requerido para la extracción por la siguiente fórmula.
![]()
Donde:
%S | Porcentaje de sólidos determinado en el ensayo preliminar 1 (Determinación preliminar del porcentaje de sólidos). |
Wf | Peso del desecho filtrado inicial o peso de la muestra total para 100% sólidos. |
-- Lentamente adicionar esta cantidad al contenedor de extracción, cerrar de forma ajustada (se recomienda usar tapas de teflón para asegurar el sello) y asegurar en el mecanismo de agitación rotatorio.
-- Se debe realizar el proceso de extracción con un blanco (usando el mismo fluido de extracción usado para las muestras) como mínimo para cada 20 extracciones que se realicen en el recipiente de extracción.
-- Rotar a 30 ± 2 rpm por 18 ± 2 horas, la temperatura del laboratorio donde se realiza la extracción debe mantenerse a 23 ± 2 ºC durante este período[20].
-- Después de las 18 ± 2 horas de extracción, separar el material dentro del recipiente de extracción en sus componentes fases líquida y sólida por filtración a través de un nuevo filtro fibra de vidrio (en este caso por bomba de vacío). En esta filtración final el papel fibra de vidrio puede ser cambiado, si se requiere para facilitar la filtración. Los filtros deben ser enjuagados con ácido si se va a evaluar la movilidad de los metales.
VI-ii.1.4 Extracto TCLP
-- Si el desecho no contenía fase líquida inicial, el líquido filtrado después de 1.3 es el “Extracto TCLP”.
-- Si el desecho contiene fase liquida y sólidos <0,5% el filtrado obtenido en 1.1 Filtración es el “Extracto TCLP”
-- Si el desecho contiene fase liquida y sólidos > 0,5%, verificar si el líquido obtenido en 1.1 Filtración “fase líquida” y el filtrado en 1.3. Extracción de la fase sólida son compatibles, combinarlos y este el “Extracto TCLP”. Si no lo son, registrar el volumen de cada uno y analizarlos por separado. Ambos son “Extracto TCLP” y al final combinar matemáticamente sus resultados:
![]()
Donde:
V1 = Volumen primera fase
C1 = Concentración primera fase
V2 = Volumen segunda fase
C2 = Concentración segunda fase.
-- Registrar el pH del Extracto inmediatamente después de su colección, tomar alícuotas para los diferentes análisis a realizar y preservar para su posterior análisis.
-- Las alícuotas para metales deben ser acidificadas con HNO3 a pH < 2. (si se observa precipitación tomar otra alícuota para análisis de metales, no acidificar, refrigerar a 4 ºC y analizar lo antes posible). Estas alícuotas deben ser digeridas en ácido excepto en aquellas circunstancias donde la digestión causa pérdidas de analitos metálicos. Si un análisis de un extracto sin digerir muestra que la concentración del analito metálico regulado excede el nivel regulatorio, el desecho se clasifica como peligroso y no se requiere digestión del extracto. Sin embargo los datos de extractos sin digerir no pueden ser usados para demostrar que el desecho no es peligroso.
-- Las otras alícuotas deben ser almacenadas bajo refrigeración 4 ºC hasta su análisis.
-- El extracto TCLP debe ser preparado y analizado acorde al método analítico apropiado.
VI-ii.2 Procedimiento para extracción de compuestos volátiles
Para obtener el extracto TCLP para compuestos volátiles solo se debe usar el dispositivo ZHE. El extracto obtenido del uso de este equipo no debe ser usado para evaluar la movilidad de analitos no volátiles (p.e. metales, pesticidas, etc.).
El equipo ZHE tiene aproximadamente 500 ml de capacidad interna. De esta forma puede acomodar como máximo 25 gramos de sólidos (definidos como la fracción de muestra para la cual no se puede forzar fuera líquido adicional, al aplicar una presión de 50 PSI), debido a la necesidad de adicionar una cantidad de fluido de extracción igual a 20 veces el peso de la fase sólida.
Recomendaciones
-- Cargar el ZHE con la muestra solo una vez y no abrir el equipo hasta que se haya recolectado el extracto final. No se permite el rellenar el ZHE para obtener 25 gramos de sólidos. No permitir que el desecho, la fase líquida inicial o el extracto estén expuestos a la atmósfera por más del tiempo que sea absolutamente necesario. Cualquier manipulación de estos materiales debe ser hecha cuando estén fríos 4 ºC para minimizar las perdidas de volátiles.
-- Si el desecho es 100% sólido pesar una submuestra de 25 gramos como máximo del desecho, registrar el peso y proceder con la preparación del equipo y materiales. En este caso no se requiere del paso de filtración de la fase líquida inicial.
-- Si el desecho contiene < 0,5% de sólidos secos la porción líquida del desecho, después de la filtración, se define como el extracto TCLP. Se debe filtrar bastante muestra de forma que la cantidad de líquido filtrado sea suficiente para todos los análisis de compuestos volátiles requeridos.
-- Para desechos conteniendo = 0,5% de sólidos secos, usar la información del porcentaje de sólidos para determinar la cantidad de muestra óptima para cargar dentro del ZHE. Los tamaños de muestra recomendados son los siguientes:
– Para desechos conteniendo < 5% de sólidos, pesar 500 gramos de sub-muestra y registrar el peso.
– Para desechos conteniendo = 5% de sólidos, determinar la cantidad de desecho a cargar dentro del ZHE como sigue:
![]()
– Pesar la submuestra del desecho del tamaño apropiado y registrar el peso.
-- Si se requiere reducción de tamaño de partícula de la porción sólida del desecho, preparar el desecho para la extracción machacando, cortando o moliendo la porción sólida del desecho a un área superficial o tamaño de partícula requerido. El desecho y el equipo de reducción apropiado deben ser refrigerados si es posible a 4 ºC antes de la reducción de tamaño de partícula. El equipo o medio usado para reducir el tamaño de las partículas no debe generar calor interna o externamente. Si se requiere reducción de la fase sólida, la exposición del desecho a la atmósfera debe ser evitada en la medida de lo posible[21].
-- Para desechos lodosos no es necesario permitir la quietud para que la fase sólida se sedimente. No se debe centrifugar los desechos antes de la filtración.
-- Se debe analizar como mínimo de un blanco usando el fluido de extracción No 1 cada 20 extracciones que se realicen.
VI-ii. 2.1 Preparación del Equipo y Materiales del ZHE
-- Prepesar el contenedor de recolección del filtrado el cual puede ser bolsa Tedlar® o jeringa dependiendo de las características del desecho como se mencionó en el numeral IV.
-- Colocar el pistón del ZHE dentro del cuerpo del equipo (como ayuda primero humedecer el empaque del pistón con el fluido de extracción). Ajustar el pistón dentro del cuerpo del ZHE hasta una altura en la cual se minimice la distancia que el pistón tenga que moverse una vez el ZHE esté cargado con la muestra (basado sobre los requerimientos de tamaño de la muestra). Asegurar el flanche de entrada/salida del gas (fondo del flanche) sobre el cuerpo del ZHE de acuerdo con las instrucciones del fabricante. Asegurar el filtro de fibra de vidrio entre las pantallas de soporte y el lado. Fijar el flanche de entrada/salida del líquido (arriba del flanche) a un lado.
-- Pesar la cantidad de muestra requerida para el análisis de acuerdo a las recomendaciones anteriores.
-- Transferir cuantitativamente la muestra completa (fase líquida y sólida) lentamente al ZHE. Asegurar todos los accesorios y colocar el mecanismo en posición vertical (flanche de entrada/salida del gas en el fondo). No atar el dispositivo de recolección del extracto al plato superior. Si el material de desecho (>1% del peso de la muestra original) está adherido al contenedor usado para transferir la muestra al ZHE, determinar el peso de este residuo y restarlo del peso de la muestra para determinar el peso del desecho que será filtrado.
-- Atar la línea de gas de la válvula de entrada/salida de gas (flanche inferior) y con la válvula de entrada/salida del líquido abierta, comenzar a aplicar lentamente presión de 1 a 10 PSI (o más si es necesario) para forzar todo el headspace lentamente fuera del mecanismo de ZHE bajo una cabina. Con la primera aparición de líquido de la válvula de entrada/salida de líquido, rápidamente cerrar la válvula y dejar de aplicar presión. Si la filtración del desecho a 4 ºC reduce la cantidad de líquido comparado con la cantidad de líquido a temperatura del cuarto, se debe permitir que la muestra se caliente hasta temperatura del cuarto en el equipo antes de la filtración. Si el desecho es 100% sólido, lentamente incrementar la presión hasta un máximo de 50 PSI para forzar más del headspace fuera del equipo.
VI-ii.2.2 Filtración fase líquida inicial
-- Atar el dispositivo de recolección del filtrado pre-pesado a la válvula de entrada/salida de líquido y abrir la válvula. Comenzar a aplicar lentamente presión de 1 a 10 PSI para forzar a la fase líquida de la muestra dentro del dispositivo. Si no se observa líquido adicional pasar a través del filtro en un período de 2 minutos, lentamente incrementar la presión en incrementos de 10 PSI hasta un máximo de 50 PSI. Después de cada incremento de 10 PSI, si no se observa líquido adicional pasar a través del filtro en un período de 2 minutos, proceder al próximo incremento de 10 PSI. Cuando el flujo del líquido ha cesado tal que la filtración a 50 PSI no resulta en filtrado adicional en 2 minutos, se debe parar la filtración. Cerrar la válvula de entrada/salida del líquido, descargar la presión del pistón y desconectar y pesar el dispositivo de recolección del filtrado[22].
-- El material en el ZHE se define como la fase sólida del desecho y el filtrado es definido como la fase líquida. La fase líquida puede ser analizada inmediatamente o almacenada a 4 ºC bajo condiciones de mínimo headspace hasta el momento de análisis[23].
VI-ii.2.3 Determinación de la cantidad de fluido de extracción
-- Determinar el peso del fluido de extracción No 1 para adicionar al equipo ZHE de la siguiente forma:
VI-ii.2.4 Llenado del equipo con el fluido de extracción
-- Con el ZHE en posición vertical, unir una línea del contenedor del fluido de extracción a la válvula de entrada/salida de líquido. La línea usada debe contener fluido de extracción fresco y debe ser prenivelado con el fluido para eliminar cualquier burbuja de aire en la línea. Liberar la presión de gas sobre el pistón del ZHE (desde la válvula de entrada/salida del gas), abrir la válvula de entrada/salida del líquido y comenzar la transferencia del fluido de extracción (por bombeo o un medio similar) dentro del ZHE. Continuar con el bombeo hasta que la cantidad apropiada del fluido sea introducida.
-- Inmediatamente después de que ha sido adicionado el fluido de extracción, cerrar la válvula de entrada/salida de líquido y desconectar la línea del fluido de extracción. Verificar el ZHE para asegurar que todas las válvulas estén en sus posiciones de cerrado. Manualmente rotar el equipo con 2 o 3 veces. Reposicionar el ZHE en la posición vertical con la válvula de entrada/salida del líquido hacia arriba. Presurizar el ZHE a 5-10 PSI (si es necesario) y lentamente abrir la válvula de entrada/salida del líquido para dejar salir cualquier headspace (dentro de una cabina) que pudo haberse introducido debido a la adición del fluido de extracción. Esta abertura debe hacerse rápidamente y debe parar con la primera aparición de líquido en la válvula. Represurizar el ZHE de 5 a 10 PSI y verificar que todos los accesorios del ZHE para asegurar que están cerrados.
VI-ii.2.5 Proceso de extracción
-- Localizar el ZHE en el aparato de agitación rotatorio y rotar a 30 ± 2 rpm por 18 ± 2 horas. La temperatura ambiente (p.e. temperatura del cuarto en el cual se realiza la extracción) debe mantenerse a 23 ± 2 ºC durante la agitación.
-- Finalizado el período de agitación de 18 ± 2 horas, verificar la presión dentro del pistón del ZHE rápidamente abriendo y cerrando la válvula de entrada/salida del gas y notar el escape de gas. Si la presión no se ha mantenido (p.e. no se observa liberación de gas), el equipo presenta una fuga. Se debe realizar el proceso de verificación de fugas presentado en el numeral IV (Equipos y Materiales), y realizar el proceso de extracción de nuevo con una nueva muestra del desecho. Si la presión dentro del equipo se ha mantenido, el material en el recipiente de extracción es separado en sus fases líquida y sólida.
VI-ii.2.6 Filtración del extracto
-- Filtrar a través del filtro de fibra de vidrio, usando el mismo proceso de filtración discutido anteriormente para ZHE de la fase líquida inicial. Si el desecho contenía una fase líquida inicial, el líquido puede ser filtrado directamente dentro del mismo dispositivo de recolección de filtrado que contenía la fase líquida inicial del desecho. Si la combinación de fluidos puede crear múltiples fases, debe usarse un dispositivo de recolección de filtrado separado, o si no hay suficiente volumen dentro del dispositivo de recolección de filtrado.
-- Todo el extracto debe ser filtrado y recolectado en los casos en los cuales se usa bolsa Tedlar®, si el extracto es de múltiples fases, o si el desecho contenía una fase líquida inicial. Un filtro fibra de vidrio en línea puede ser usado para filtrar el material dentro del ZHE si se sospecha que el filtro dentro del equipo se ha roto.
VI-ii.3 Extractos TCLP
-- Si el desecho original no contenía una fase líquida inicial, el líquido filtrado del extracto es definido como el Extracto TCLP.
-- Si el desecho contenía una fase líquida inicial, el material líquido filtrado obtenido después del proceso de extracción y la fase líquida inicial se definen colectivamente como el extracto TCLP.
-- Si el desecho no contenía sólidos o los sólidos secos eran <0,5 % la fase líquida inicial corresponde al Extracto TCLP.
Preparación de extracto para análisis.
-- Inmediatamente después de la recolección del extracto TCLP, preparar el extracto para análisis y almacenar con el mínimo espacio de cabeza a 4 ºC hasta su análisis. Se debe realizar el análisis del Extracto TCLP de acuerdo al método analítico apropiado (Ver Anexo 5).
-- Si las fases individuales van a ser analizadas por separado (p.e. que no sean miscibles), determinar el volumen de las fases individuales (a 0,5 %), realizar los análisis por separado y combinar los resultados matemáticamente usando un promedio ponderado por volumen.
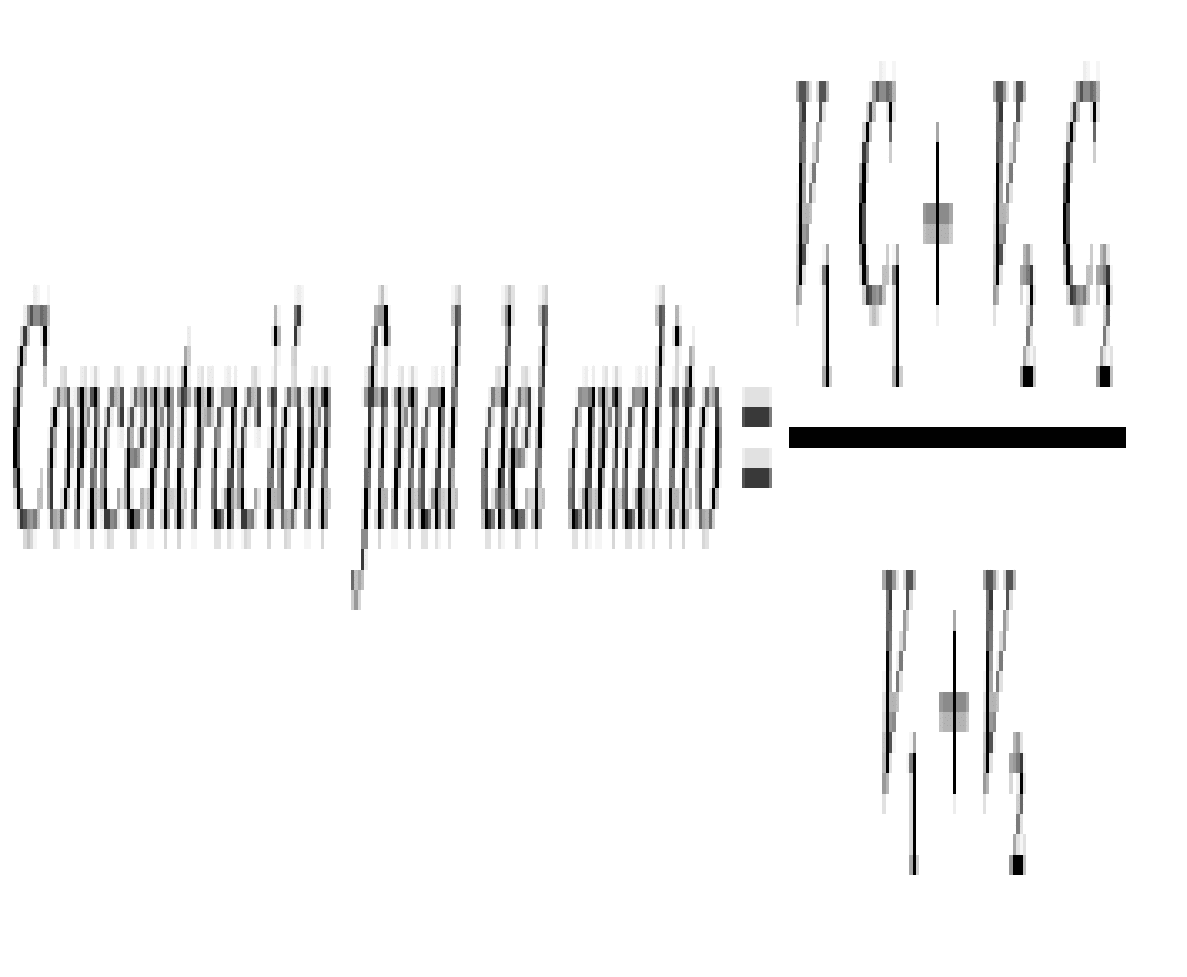
Donde:
V1 | Volumen primera fase (L) |
C1 | Concentración del analito en la primera fase (mg/l) |
V2 | Volumen segunda fase (L) |
C2 | Concentración del analito en la segunda fase (mg/l) |
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 6.1. Diagrama de Flujo – Evaluaciones preliminares.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 6.2. Diagrama de flujo – Ensayo para compuestos no-volátiles.
VII. RESULTADOS
Este método presenta el proceso de obtención del Extracto TCLP. Obtenido este se deben realizar las determinaciones para cada analito de interés siguiendo los métodos analíticos adecuados (Anexo 5). Comparar las concentraciones de los analitos en el extracto TCLP con los niveles identificados en la regulación apropiada.
VIII. ASEGURAMIENTO DE LA CALIDAD
-- Se debe analizar como mínimo un blanco (usando el mismo fluido de extracción usado para las muestras) cada 20 extracciones realizadas.
-- Se debe realizar un ensayo con matriz dopada para cada tipo de desecho (p.e. lodos de plantas de tratamiento de aguas residuales, suelos contaminados, etc.) a menos que los resultados excedan el nivel regulatorio y los datos estén siendo usados únicamente para demostrar que el desecho excede el nivel regulatorio. Se requiere que como mínimo una matriz dopada sea analizada para cada bache de análisis. Como mínimo, seguir la guía de adición para matriz estándar de cada método analítico.
– Los dopajes a la matriz se adicionan después de la filtración del Extracto TCLP y antes de la preservación. Los dopajes no deben ser adicionados antes de la extracción de la muestra.
– En la mayoría de los casos, los dopajes deben ser adicionados a una concentración equivalente al nivel regulatorio correspondiente. Si la concentración del analito es menor que la mitad del nivel regulatorio, la concentración de dopaje puede ser tan bajo como la mitad de la concentración del analito, pero no debe ser menor que cinco veces el límite de detección del método. Para evitar diferencias en el efecto matriz, los dopajes deben ser adicionados al mismo volumen nominal del Extracto TCLP del que fue analizado en la matriz sin dopar.
– El propósito del dopaje es monitorear el comportamiento del método analítico usado, y determinar si existen interferencias de matriz. Cuando la recuperación de la matriz dopada es por debajo del comportamiento del método analítico esperado, se deben usar otros métodos de calibración interna, modificaciones de los métodos analíticos, o uso de métodos analíticos alternativos de forma que se alcance una medición confiable de la concentración del analito.
– El porcentaje de recuperación de matrices dopadas son calculados por la siguiente fórmula:
![]()
Donde:
Xs | valor medido para la muestra dopada |
Xu | valor medido para la muestra sin dopaje |
K | valor conocido de dopaje en la muestra. |
-- Todas las mediciones de control de calidad descritos en los métodos analíticos apropiados se deben seguir.
-- El uso de métodos de cuantificación de calibración interna deben ser empleados para contaminantes metálicos si, a) la recuperación del contaminante desde el extracto TCLP es menor de 50% y la concentración no excede el nivel regulatorio o, b) la concentración del contaminante medido en el extracto está dentro del 20% del nivel regulatorio apropiado.
– El método de adiciones estándares debe ser empleado como un método de cuantificación de calibración interna para cada uno de los contaminantes metálicos.
– El método de adiciones estándares requiere la preparación de estándares de calibración en la matriz de la muestra en vez de prepararlos en agua grado reactivo o blanco de la solución. Se requiere tomar 4 alícuotas idénticas de la solución y adicionar cantidades conocidas de estándar a 3 de estas alícuotas. La cuarta alícuota es la desconocida. Preferiblemente, la primera adición debe ser preparada de forma tal que la concentración resultante sea aproximadamente el 50% de la concentración esperada de la muestra. La segunda y tercera adición deben ser preparadas de forma tal que las concentraciones sean aproximadamente el 100 y el 150% de la concentración esperada de la muestra. Las cuatro alícuotas son llevadas al mismo volumen final por adición de agua grado reactivo o una solución de blanco, y pueden necesitar ajuste de dilución para mantener la señal en el rango lineal de la técnica del instrumento. Se analizan las cuatro alícuotas.
– Graficar la señal del instrumento o concentración derivada de la calibración externa como la variable dependiente (eje y) versus la concentración de las adiciones de estándar como la variable independiente (eje x). Determinar el intercepto de la abscisa (variable independiente, eje x) la cual será la concentración desconocida.
– Alternativamente, restar la señal del instrumento o la concentración derivada de la calibración externa de la matriz desconocida de la señal o concentración derivada de la calibración externa de las adiciones estándares. Graficar o hacer una regresión lineal de la señal del instrumento corregido o de la concentración derivada de la calibración externa corregida como variable dependiente versus la variable independiente. Calcular las concentraciones para las concentraciones desconocidas en la curva de calibración interna como si fuera una curva de calibración externa.
-- La siguiente tabla presenta los períodos de tiempo de mantenimiento máximo de la muestra entre las diferentes etapas del procesamiento para el análisis de TCLP
Tiempo máximo de mantenimiento de muestra (días)
Desde | Toma de muestra | Extracción | Preparación del extracto | Tiempo total gastado |
Hasta | Extracción | Preparación del extracto | Análisis | |
Compuestos volátiles | 14 | NA | 14 | 28 |
Compuestos semivolátiles | 14 | 7 | 40 | 61 |
Mercurio | 28 | NA | 28 | 56 |
Metales (Excepto mercurio) | 180 | NA | 180 | 360 |
Si los tiempos de mantenimiento son excedidos, los valores obtenidos pueden ser considerados como concentraciones mínimas. Si se excede el tiempo de mantenimiento no es aceptable para establecer si el desecho no excede el nivel regulatorio. Si se excede el tiempo de mantenimiento no invalida la caracterización del desecho si este excede el nivel regulatorio.
IX. INFORMACION ESTADISTICA
IX-i. ROBUSTEZ
La EPA ha realizado dos estudios de robustez para determinar el efecto de varias perturbaciones sobre los elementos específicos del protocolo de TCLP. Los ensayos de robustez determinan la sensibilidad de variaciones pequeñas en el procedimiento, los cuales pueden ser esperados que ocurran durante la aplicación rutinaria del laboratorio.
IX-i.1 Metales
Las siguientes condiciones fueron usadas cuando se realizó el proceso de extracción de un desecho para el análisis de metales:
| Variación en las condiciones del ensayo | |
| Relación líquido/sólido | 19:1 vs 21:1 |
| Tiempo de extracción | 16 horas vs 18 horas |
| Espacio de cabeza | 20 vs 60% |
| Acidez del buffer No. 2 | 190 meq vs 210 meq |
| Filtros enjuagados con ácido | Si vs no |
| Tipo de filtro | Fibra de vidrio 0,7 ìm vs 0,45 ìm vs policarbonato |
| Tipo de botellas | Borosilicato vs vidrio cristalería |
De las siete variaciones del método evaluadas, la acidez del fluido de extracción tuvo el más grande impacto sobre los resultados. Cuatro de los 13 metales de un desecho de una mezcla de desecho de industria galvánica y un lodo de un separador API y dos de tres metales de un desecho del fondo de una excavación de soda de amonio fueron extraídos a niveles más altos por el buffer de mayor acidez. Debido a la sensibilidad a los cambios de pH, el método requiere que los fluidos de extracción sean preparados de forma tal que el pH final esté dentro de ± 0,05 unidades como lo especifica el protocolo.
IX-i.2 Compuestos orgánicos volátiles
Las siguientes condiciones fueron usadas por la EPA cuando se extrajo un desecho para análisis de compuestos orgánicos volátiles:
| Variación en las condiciones del ensayo | |
| Relación líquido/sólido | 19:1 vs 21:1 |
| Espacio de cabeza | 0% vs 5% |
| Acidez del buffer No. 1 | 60 meq vs 80 meq |
| Método de almacenar el extracto | Jeringa vs Bolsa Tedlar® |
| Alícuotas | Si vs no |
| Presión dentro del pistón | 0 PSI vs 20 PSI |
Ninguno de los parámetros tuvo un efecto significativo sobre los resultados del ensayo de robustez.
IX-ii. Precisión
Se han realizado muchos estudios de precisión (reproducibilidad) de TCLP, y han mostrado que, en general, la precisión del TCLP es comparable a, o excede la del ensayo de toxicidad EP (SW-846 1310) y que la precisión del método es adecuada. Una de las contribuciones más significativas para la adecuada precisión parece estar relacionada a la homogeneidad de la muestra y las variaciones interlaboratorios (debido a la naturaleza de los materiales de desecho).
IX-ii.1 Metales
Los resultados de estudios multilaboratorios realizados por la EPA se muestran en la siguiente tabla e indican que un análisis sencillo de un desecho puede no ser adecuado para la caracterización del desecho y los requerimientos de identificación.
| Desecho | Fluido de extracción | metal | X | S | % RSD |
| Fondos de extracción soda amonio | 1 | Cadmio | 0,053 | 0,031 | 60 |
| 2 | 0,023 | 0,017 | 76 | ||
| 1 | Cromo | 0,015 | 0,0014 | 93 | |
| 2 | 0,0032 | 0,0037 | 118 | ||
| 1 | Plomo | 0,0030 | 0,0027 | 90 | |
| 2 | 0,0032 | 0,0028 | 87 | ||
| Mezcla de API/EW | 1 | Cadmio | 0,0046 | 0,0028 | 61 |
| 2 | 0,0005 | 0,0004 | 77 | ||
| 1 | Cromo | 0,0561 | 0,0227 | 40 | |
| 2 | 0,105 | 0,018 | 17 | ||
| 1 | Plomo | 0,0031 | 0,0031 | 100 | |
| 2 | 0,0124 | 0,0136 | 110 | ||
| Cenizas de combustible fósil | 1 | Cadmio | 0,080 | 0,069 | 86 |
| 2 | 0,093 | 0,067 | 72 | ||
| 1 | Cromo | 0,017 | 0,014 | 85 | |
| 2 | 0,070 | 0,040 | 57 | ||
| 1 | Plomo | 0,0087 | 0,0074 | 85 | |
| 2 | 0,0457 | 0,0083 | 18 | ||
Nota: X media de los resultados de entre 6 y 12 diferentes laboratorios.
Unidades en mg/l, fluido de extracción No 1 pH = 4,9 y No 2 pH= 2,9.
IX-ii.2 Compuestos orgánicos semivolátiles
Los resultados de dos estudios realizados por la EPA se muestran en las siguientes tablas. La precisión de laboratorio fue más del 90%. Los resultados presentaron una desviación estándar relativa menor del 25%. Más del 85% de los compuestos individuales en los estudios multilaboratorios estuvieron dentro del rango de desviación estándar relativa de 20 al 120%. Ambos estudios concluyeron que los TCLP suministran precisión adecuada. Se determinó que un alto contenido de acetato en el fluido de extracción no presentó problemas para las condiciones analíticas usadas (p.e., degradación de la columna del cromatógrafo de gases).
Desecho | Compuesto | Fluido de extracción | X | S | % RSD |
Fondos de excavación de soda amonio | Fenol | 1 | 19000 | 2230 | 11,6 |
| 2 | 19400 | 929 | 4,8 | ||
| 2-metilfenol | 1 | 2000 | 297 | 14,9 | |
| 2 | 1860 | 52,9 | 2,8 | ||
| 4-metilfenol | 1 | 7940 | 1380 | 17,4 | |
| 2 | 7490 | 200 | 2,7 | ||
| 2,4-Dimetilfenol | 1 | 321 | 46,8 | 14,6 | |
| 2 | 307 | 45,8 | 14,9 | ||
| Naftaleno | 1 | 3920 | 413 | 10,5 | |
| 2 | 3827 | 176 | 4,6 | ||
| 2-metilnaftaleno | 1 | 290 | 44,8 | 15,5 | |
| 2 | 273 | 19,3 | 7,1 | ||
| Dibenzofurano | 1 | 187 | 22,7 | 12,1 | |
| 2 | 187 | 7,2 | 3,9 | ||
| Acenaftileno | 1 | 703 | 89,2 | 12,7 | |
| 2 | 663 | 20,1 | 3,0 | ||
| Fluoreno | 1 | 151 | 17,6 | 11,7 | |
| 2 | 156 | 2,1 | 1,3 | ||
| Fenantreno | 1 | 241 | 22,7 | 9,4 | |
| 2 | 243 | 7,9 | 3,3 | ||
| Antraceno | 1 | 33,2 | 6,19 | 18,6 | |
| 2 | 34,6 | 1,55 | 4,5 | ||
| Fluorantreno | 1 | 25,3 | 1,8 | 7,1 | |
| Mezcla de API/EW | 2 | 26,0 | 1,8 | 7,1 | |
| Fenol | 1 | 40,7 | 13,5 | 33,0 | |
| 2 | 19,0 | 1,76 | 9,3 | ||
| 2,4 Dimelfenol | 1 | 33,0 | 9,35 | 28,3 | |
| 2 | 43,3 | 8,61 | 19,9 | ||
| Naftaleno | 1 | 185 | 29,4 | 15,8 | |
| 2 | 165 | 24,8 | 15,0 | ||
| 2-metilnaftaleno | 1 | 265 | 61,2 | 23,1 | |
| 2 | 200 | 18,9 | 9,5 | ||
Rango de RSD% = 1-33
Media de RSD% = 12
Nota: unidades = íg/L
Las extracciones fueron realizadas por triplicado, todos los resultados fueron al menos 2x el límite de detección, fluido de extracción No 1 = pH = 4,9, fluido de extracción No 2 pH= 2,9
| Desecho | Compuesto | Fluido de extracción | X | S | % RSD |
| Fondos de excavación soda amonio (A) | BNA | 1 | 10043 | 7680 | 76,5 |
| 2 | 10376 | 6552 | 63,1 | ||
| Mezcla de API/EW (B) | BNA | 1 | 1624 | 675 | 41,6 |
| 2 | 2074 | 1463 | 70,5 | ||
| Ceniza de combustible fósil (C) | BNA | 1 | 750 | 175 | 23,4 |
| 2 | 739 | 342 | 46,3 | ||
Media de RSD% = 54
Nota: unidades = íg/L
X = media de resultados de 3 a 10 laboratorios
Fluido de extracción No 1 = pH = 4,9, fluido de extracción No 2 pH= 2,9Rango de RSD% para compuestos individuales
A, número1-0-113 / A, número2-28-108 / B, número1-20-156 / B, número2-49-128 / C, número1-36-143 / C, número2-61-164
IX-ii.3 Compuestos orgánicos volátiles
En un estudio realizado por la EPA en el cual participaron 11 laboratorios se evaluaron dos tipos de desechos los cuales fueron dopados con una mezcla de COV. Los resultados del estudio se presentan en la siguiente tabla. La precisión de los resultados tiende a cubrir un rango considerable. Sin embargo, el rango y la media de la desviación estándar relativa son comparables con los resultados del estudio de metales presentados en el numeral 1. Los autores del estudio concluyeron que a un nivel de significancia del 95%:
1. Las recuperaciones entre laboratorios fueron estadísticamente similares.
2. Las recuperaciones no variaron significativamente entre dos tipos de muestra, y
3. Cada laboratorio mostró el mismo patrón de recuperación para cada una de las dos muestras.
| Desecho | Compuesto | X | S | % RSD |
| Sobrantes de Minas | Cloruro de vinilo | 6,36 | 6,36 | 100 |
| Cloruro de metileno | 12,1 | 11,8 | 98 | |
| Disulfuro de carbono | 5,57 | 2,83 | 51 | |
| 1,1-dicloroeteno | 21,9 | 27,7 | 127 | |
| 1,1-dicloroetano | 31,4 | 25,4 | 81 | |
| Cloroformo | 46,6 | 29,2 | 63 | |
| 1, 2 -dicloroetano | 47,8 | 33,6 | 70 | |
| 2-butano | 43,5 | 36,9 | 85 | |
| 1, 1, 1- tricloroetano | 20,9 | 20,9 | 100 | |
| Tetracloruro de carbono | 12,0 | 8,2 | 68 | |
| Tricloroeteno | 24,7 | 21,2 | 86 | |
| 1,1, 2- tricloroeteno | 19,6 | 10,9 | 56 | |
| Benceno | 37,9 | 28,7 | 76 | |
| 1, 1, 2, 2-tetracloroetano | 34,9 | 25,6 | 73 | |
| Tolueno | 29,3 | 11,2 | 38 | |
| Clorobenceno | 35,6 | 19,3 | 54 | |
| Etilbenceno | 4,27 | 2,80 | 66 | |
| Triclorofluorometano | 3,82 | 4,40 | 115 | |
| Acrilonitrilo | 76,7 | 110,8 | 144 | |
| Fondos de excavación soda amonio | Cloruro de vinilo | 5,00 | 4,71 | 94 |
| Cloruro de metileno | 14,3 | 13,1 | 92 | |
| Disulfuro de carbono | 3,37 | 2,07 | 61 | |
| 1,1-dicloroeteno | 52,1 | 38,8 | 75 | |
| 1,1-dicloroetano | 52,8 | 25,6 | 49 | |
| Cloroformo | 64,7 | 28,4 | 44 | |
| 1,2 – dicloroetano | 43,1 | 31,5 | 73 | |
| 2-butano | 59,0 | 39,6 | 67 | |
| 1,1,1 – tricloroetano | 53,6 | 40,9 | 76 | |
| Tetracloruro de carbono | 7,10 | 6,1 | 86 | |
| Tricloroeteno | 57,3 | 34,2 | 60 | |
| 1,1,2 – tricloroeteno | 6,7 | 4,7 | 70 | |
| Benceno | 61,3 | 26,8 | 44 | |
| 1,1,2,2 – tetracloroetano | 3,16 | 2,1 | 66 | |
| Tolueno | 69,0 | 18,5 | 27 | |
| Clorobenceno | 71,8 | 12,0 | 17 | |
| Etilbenceno | 3,70 | 2,2 | 58 | |
| Triclorofluorometano | 4,05 | 4,8 | 119 | |
| Acrilonitrilo | 29,4 | 34,8 | 118 | |
Rango RSD% = 17 – 144
Media RSD% = 75
Nota: unidades = íg/L
XI. REFERENCIAS
Método 1311. Procedimiento de lixiviación para la característica de toxicidad. SW 846. Revisión 0 julio 1992
6.2 Procedimiento de lixiviación de precipitación sintética-SPLP
I. ALCANCE Y APLICABILIDAD
-- Este método presenta el procedimiento para la obtención de un extracto SPLP. No presenta los métodos de ensayo de laboratorio para cada compuesto individual.
-- Este método está diseñado para determinar la movilidad de los analitos orgánicos e inorgánicos presentes en un desecho líquido, sólido o de múltiples fases.
II. PRINCIPIO DEL METODO
i) Desechos líquidos (aquellos que contienen menos de 0,5% de sólidos secos).
El Extracto SPLP es la fase líquida obtenida después de la filtración del desecho a través de un papel fibra de vidrio de 0,6 a 0,8 µm.;
ii) Desechos conteniendo mayor o igual cantidad de 0,5% de sólidos.
El desecho es filtrado y el líquido filtrado corresponde a extracto SPLP fase líquida y se almacena. La fase sólida del desecho se reduce de tamaño (si es requerido), y se realiza un proceso de extracción al ponerla en contacto con un fluido de extracción igual a 20 veces el peso de la fase sólida durante 18 ± 2 horas a 30 ± 2 rpm. Después del período de extracción, la fase sólida con el fluido son filtrados separando el extracto líquido. Si la fase líquida y el extracto líquido son compatibles, estas se mezclan y se analizan juntas. Si son incompatibles los líquidos son analizados por separado y los resultados son combinados matemáticamente para obtener una concentración promedia ponderada por volumen;
iii) Desechos 100% sólidos.
El desecho se reduce de tamaño (si es requerido), y se realiza un proceso de extracción al ponerla en contacto con un fluido de extracción igual a 20 veces su peso seco durante 18 ± 2 horas a 30 ± 2 rpm. Después del período de extracción la fase sólida con el fluido son filtradas separando el extracto líquido. El extracto líquido es el Extracto SPLP y se analiza.
III. INTERFERENCIAS
Interferencias potenciales que pueden ser encontradas durante el análisis deberán ser revisadas en los métodos analíticos individuales.
IV. EQUIPOS Y MATERIALES
-- Aparato de agitación
Este debe ser capaz de rotar los recipientes de extracción en rotación completa a 30 ± 2 rpm (Ver Anexo 2).
-- Recipientes de extracción:
– Recipiente de extracción de cero espacio de cabeza – (zero-headspace extraction vessel) ZHE (Ver anexo 3-2 TCLP).
Este se usa solo cuando se desea evaluar la movilidad de analitos volátiles en el desecho. El ZHE permite la separación líquido/sólido dentro del equipo, excluyendo el espacio de cabeza. Este tipo de recipiente permite la separación líquido/sólido inicial, extracción, y filtración del extracto final sin abrir el recipiente. Este recipiente debe tener un volumen interno de 500 a 600 ml, y estar equipado para acomodar un filtro de 90 a 110 mm. El recipiente contiene empaques de VITON los cuales deben ser reemplazados frecuentemente. En el Anexo 4 se presentan los recipientes ZHE conocidos por la EPA.
Para que el ZHE sea aceptado, el pistón interno debe ser capaz de moverse aproximadamente a 15 PSI o menos. Si se requiere más presión los empaques deben ser reemplazados, si con esto no se resuelve el problema, el ZHE no se acepta para el análisis de SPLP y el fabricante debe ser contactado.
Después de cada extracción debe verificarse que no presente fugas, si el equipo tiene un medidor de presión, se debe presurizar hasta 50 PSI, dejar quieto por 1 hora, sumergir en agua y verificar la presencia de burbujas de aire escapando de cualquier accesorio. Si se pierde la presión verificar todos los accesorios e inspeccionar y reemplazar los empaques, si es necesario. Reensayar el mecanismo. Si los problemas de fugas no se pueden resolver, se debe contactar al fabricante.
– Recipientes de extracción de botella
Si se va avaluar compuestos no volátiles en el desecho, se requiere un contenedor con suficiente capacidad para retener la muestra y el fluido de extracción. En este se permite espacio de cabeza.
Las botellas de extracción pueden ser de diferentes materiales, dependiendo de los analitos a ser analizados y la naturaleza del desecho. Se recomienda usar botellas de vidrio, botellas plásticas de politetrafluoroetileno (PTFE) cuando se va a analizar tanto compuestos inorgánicos y orgánicos. Si solo se van a analizar metales se pueden usar recipientes de polietileno de alta densidad (HDPE), polipropileno (PP) o cloruro de polivinilo (PVC), y se recomiendan botellas de vidrio de borosilicato.
-- Equipos de filtración (se recomienda que todas las filtraciones sean realizadas bajo campana de extracción)
– Recipiente de extracción de cero espacio de cabeza ZHE
Cuando se vayan a analizar compuestos volátiles se debe usar este equipo para la filtración. El equipo debe ser capaz de soportar y mantener en su lugar el papel de filtro fibra de vidrio y ser capaz de resistir la presión necesaria para lograr la separación (50 PSI).
Cuando se sospeche que el papel filtro fibra de vidrio se ha roto, un papel filtro fibra de vidrio en línea puede ser usado para filtrar el material dentro del ZHE
– Bomba de vacío
Puede solo ser usada para filtración de desechos con un bajo contenido de sólidos (<10%) y desechos altamente granulares, cuando se van a analizar compuestos no volátiles. El equipo debe tener un volumen interno de 300 ml y permitir acomodar un papel filtro como mínimo de 47 mm (se recomiendan los retenedores de capacidad interna de 1,5 L o mayor y equipados para acomodar filtros de diámetro de 142 mm). En el anexo 4 se presentan algunos equipos conocidos de la EPA. Los materiales de los equipos pueden ser vidrio, PTFE, o acero inoxidable; si solo se van a evaluar metales se acepta HDPE, PP o PVC y se recomienda vidrio borosilicato.
– Equipos de filtración de presión positiva (capaces de alcanzar hasta 50 PSI): se usan para desechos con alto contenido de sólidos (>10%). El equipo debe tener un volumen interno de 300 ml y permitir acomodar un papel filtro como mínimo de 47 mm (se recomiendan los retenedores de capacidad interna de 1,5 L o mayor y equipados para acomodar filtros de diámetro de 142 mm). En el Anexo 4 se presentan algunos equipos conocidos de la EPA. Los materiales de los equipos pueden ser vidrio, PTFE, o acero inoxidable si solo se va avaluar metales se acepta HDPE, PP o PVC, y se recomienda vidrio borosilicato.
-- Papel filtro
Fibra de vidrio de borosilicato con un tamaño de poro efectivo entre 0,6 a 0,8 µm. Cuando se evalúa la movilidad de metales, los filtros deben ser enjuagados con ácido (enjuagar con ácido nítrico 1N seguido por tres enjuagues consecutivos de agua destilada desionizada, se recomienda como mínimo 1L) antes de usarse. Los filtros conocidos por la EPA se presentan en el Anexo 4.
-- PHmetro
Debe garantizar una exactitud de ± 0,05 unidades a 25 ºC
-- Mecanismo de recolección del Extracto ZHE: bolsas Tedlar®, o jeringas sello de gas de vidrio, acero inoxidable o PTFE. Para la selección de este tener en cuenta las siguientes especificaciones:
– Si el desecho contiene una fase líquida acuosa o si el desecho no contiene una cantidad significativa de líquido no acuoso (< 1% del desecho total), se debe usar bolsa Tedlar® o jeringa de 600 ml para recolectar y combinar el líquido inicial y el extracto del sólido.
– Si el desecho contiene una cantidad significativa de líquido no acuoso en la fase líquida inicial (> 1% del desecho total), se puede usar jeringa o bolsa Tedlar® tanto para la separación líquido sólido inicial y el extracto de la filtración final. Sin embargo se debe usar una o la otra y no ambas.
– Si el desecho no contiene fase líquida inicial (es 100% sólido) o no tiene una fase sólida significativa (es 100% líquido, o <0,5% de sólidos secos) se puede usar bolsa Tedlar® o jeringa. Si se usa jeringa descartar los primeros 5 ml del líquido obtenidos del ZHE. Las demás alícuotas son usadas para el análisis.
-- Mecanismo de transferencia del fluido de extracción al ZHE: cualquier dispositivo capaz de transferir el fluido de extracción dentro del ZHE sin cambiar la naturaleza de este (por ejemplo una bomba peristáltica o de desplazamiento positivo, una jeringa con sello, unidad de filtración de presión u otro mecanismo).
-- Balanza de laboratorio: que garantice una exactitud de ± 0,01 g.
-- Vaso de precipitado o erlenmeyer de vidrio de 500 ml.
-- Vidrio de reloj, de un diámetro apropiado para cubrir el vaso o erlenmeyer.
-- Agitador magnético.
-- Estufa o plancha de calentamiento.
-- Centrífuga (opcional como ayuda de filtración).
V. REACTIVOS
Todos los reactivos que se deben utilizar son grado reactivo a menos que se mencione explícitamente otro grado.
-- Agua grado reactivo: para analitos no volátiles agua Tipo II ASTM o equivalente. Para extracciones de compuestos volátiles se recomienda que el agua grado reactivo sea generada al pasar agua potable a través de un filtro de cama de carbón activado conteniendo alrededor de 500 gramos de este; o con un sistema de purificación (Millipore Super-Q o equivalente); o por ebullición de agua por 15 minutos, después y mientras se mantiene a 90 ± 5 ºC burbujear un gas inerte libre de contaminantes (p.ej. nitrógeno) por 1 hora, mientras esta caliente transferir a una botella de boca angosta sin espacio de cabeza y sellar con septa de teflón y tapar.
-- Acido Sulfúrico/Nítrico (Mezcla en porcentaje de peso 60/40). Cautelosamente mezclar 60 g de ácido sulfúrico concentrado con 40 g de ácido nítrico concentrado. Si se prefiere, se puede preparar con una mezcla de ácidos H2SO4/HNO3 más diluidos lo cual hace más fácil el ajuste del pH en los fluidos de extracción.
-- Fluidos de extracción
Fluido de extracción No 1.
Adicionar la mezcla de la mezcla de ácidos H2SO4/HNO3 60/40 en porcentaje a peso a agua grado reactivo hasta un pH de 4,20 ± 0,05. Este fluido es usado para determinar el grado de lixiviación de desechos[24].
Fluido de extracción No 2.
Este fluido no se utiliza para ensayo de lixiviado de desechos.
Fluido de Extracción No 3.
Este fluido es agua grado reactivo y es usada para determinar el grado de lixiviación de cianuro y compuestos volátiles[25].
-- Estándares analíticos: preparados de acuerdo al método analítico apropiado.
VI. PROCEDIMIENTO
VI-i. Evaluaciones preliminares
Se requiere una alícuota del desecho mínimo de 100 gramos. Esta no se utilizará para obtener el extracto SPLP.
VI-i.1 Determinación preliminar del porcentaje de sólidos
Este es definido como la fracción de la muestra de la cual no se puede forzar a salir más líquido por la presión aplicada, siguiendo el procedimiento descrito a continuación.
-- Para desechos 100% sólidos ir al punto 3 ensayo preliminar de reducción de tamaño.
-- Si la muestra es líquida o de múltiples fases, se requiere hacer una separación líquido/sólido.
-- Prepesar el filtro y el envase que recibirán el líquido filtrado.
-- Ensamblar el equipo de filtración y el filtro. Colocar el filtro sobre el soporte y asegurar.
-- Pesar una alícuota del desecho y registrar el peso (mínimo 100 g).
-- Permitir que la fase sólida se asiente. Si el desecho se sedimenta muy lentamente este puede ser centrifugado antes de la filtración. La centrifugación debe ser usada únicamente como una ayuda de filtración, si se usa, el líquido debe ser decantado y filtrado, y después filtrar la porción sólida a través del mismo sistema de filtración.
-- Cuantitativamente transferir la muestra del desecho al retenedor del filtro (fase líquida y sólida). Dispersar la muestra del desecho uniformemente sobre la superficie del filtro.
-- Gradualmente aplicar vacío o presión de 1 a 10 PSI, hasta que el aire o gas presurizado se mueva a través del filtro, si esto no se alcanza a los 10 PSI y/o si no se observa el paso de líquido en un período de 2 minutos, lentamente incrementar la presión en incrementos de 10 PSI hasta un máximo de 50 PSI. Cuando el gas presurizado comienza a moverse a través del filtro, o cuando el flujo del líquido ha cesado a 50 PSI se detiene la filtración.
-- El material retenido en el filtro es definido como la fase sólida del desecho y el filtrado como la fase líquida[26].
-- Determinar el peso de la fase líquida restando el peso del erlenmeyer vacío, del peso del erlenmeyer lleno con el filtrado después de la filtración.
-- Determinar el peso de la fase sólida de la muestra restando el peso de la fase líquida del peso total de la muestra, registrado anteriormente.
-- Registrar el peso de las fases líquida y sólida. Calcular el % de sólidos (%S) como sigue:
Donde:
| Ws | Peso sólido |
| Wt | Peso total de la muestra. |
-- Si el porcentaje de sólidos es menor de 0,5%, proceder al ensayo para no volátiles y volátiles donde el filtrado será el extracto SPLP.
-- En el caso en que el porcentaje de sólidos sea mayor a 0,5% y se observe que una pequeña cantidad del filtrado esta contiene agua en el filtro seguir con la determinación del porcentaje de sólidos secos, si no se observa esa pequeña cantidad de humedad pasar a la determinación de reducción de tamaño.
VI-i.2 Determinación del porcentaje de sólidos secos
-- Remover la fase sólida y el filtro del aparato de filtración.
-- Secar la fase sólida y el filtro a 100 ± 20 ºC, hasta peso constante (hasta que dos mediciones sucesivas produzcan el mismo valor dentro de ± 1%), registrar el peso final
-- Calcular el porcentaje de sólidos secos con la siguiente ecuación.
![]()
Donde:
Ws = Peso del filtro y del desecho seco
Wt = Peso inicial del desecho.
-- Si el porcentaje de sólidos secos es menor de 0,5%, entonces proceder al ensayo para no volátiles y volátiles.
-- Si el porcentaje de sólidos secos es mayor a 0,5% y si se va a realizar extracción para compuestos no volátiles iniciar el ensayo preliminar 1 con una porción fresca del desecho para determinar si se requiere reducción de tamaño de partícula, si solo se va a realizar extracción para volátiles seguir con el procedimiento para determinación del fluido de extracción.
VI-i.3 Determinación si el desecho requiere reducción de tamaño de partículas
Para desechos con un porcentaje de sólidos = 0.5 % o un desecho 100 % sólido se debe evaluar si este requiere reducción de tamaño de partículas. Este se requiere cuando los sólidos tienen un área superficial por gramo de material igual o mayor de 3,1 cm2, o si los sólidos presentan un tamaño mayor de 0,95 cm en su menor dimensión. Se puede realizar la evaluación a la vista, o en el caso de tener dudas pasar la fase sólida a través de un tamiz estándar de 9,5 mm (0,375 pulgadas), si quedan partículas retenidas se requiere que a la fase sólida de la muestra en el ensayo se le haga reducción de tamaño de partícula machacándola, cortándola, moliéndola hasta llegar al área superficial o tamaño requerido.
VI-i.4 Determinación del fluido apropiado para la extracción
-- Para desechos, usar el fluido de extracción No 1
-- Para desechos conteniendo cianuro usar el fluido de extracción No 3 (agua grado reactivo), ya que la lixiviación de muestras conteniendo cianuro bajo condiciones ácidas puede resultar en la formación de gas de cianuro de hidrógeno.
VI-ii. Ensayo
VI-ii.1 Procedimiento para extracción de compuestos no volátiles
Se requiere un tamaño de muestra como mínimo de 100 gramos (de fase sólida y líquida). En algunos casos, puede ser apropiada una gran cantidad de muestra, dependiendo del contenido de sólidos, de si la fase líquida inicial es miscible con el extracto de la fase sólida, y de la cantidad de analitos de interés (inorgánicos, compuestos orgánicos semivolátiles, pesticidas y herbicidas). En estos casos se requiere bastantes sólidos para generar suficiente volumen de extracto SPLP, si la cantidad de extracto generado en una extracción no es suficiente, se requerirá más de una extracción y los extractos deben ser combinados y de este obtener alícuotas para los diferentes análisis. Si el desecho es 100% sólido pesar una submuestra del desecho, realizar reducción de tamaño si se requiere, y luego pasar a la etapa de extracción con el fluido seleccionado.
VI-ii.1.1. Filtración
Si las muestras son líquidas o de múltiples fases, se requiere separación líquido sólido.
-- Prepesar recipiente que recibirá el filtrado.
-- Ensamblar el equipo de filtración. Colocar el filtro sobre el soporte y asegurar. Enjuagar el filtro con acido si se va a evaluar la movilidad de metales[27].
-- Pesar una alícuota del desecho y registrar el peso (mínimo 100 g).
-- Permitir que la fase sólida se asiente. Si el desecho se sedimenta muy lentamente, estos pueden ser centrifugados antes de la filtración. La centrifugación puede ser usada solo como una ayuda de filtración, si esta se usa el líquido debe ser decantado y filtrado y luego la porción sólida a través del mismo sistema de filtración.
-- Cuantitativamente transferir la muestra del desecho al retenedor del filtro (fase líquida y sólida). Dispersar la muestra del desecho uniformemente sobre la superficie del filtro[28].
-- Para muestras con un porcentaje de sólidos < 10% se puede usar bomba de vacío, para las que presentan un mayor contenido de sólidos se debe usar una bomba de presión positiva. Gradualmente aplicar vacío o presión de 1 a 10 PSI, hasta que el aire o gas presurizado se mueva a través del filtro, si esto no se alcanza a los 10 PSI y si no se observa el paso de líquido en un período de 2 minutos, lentamente incrementar la presión en incrementos de 10 PSI hasta un máximo de 50 PSI. Cuando el gas presurizado comienza a moverse a través del filtro, o cuando el flujo del líquido ha cesado a 50 PSI se detiene la filtración[29].
-- El material retenido en el filtro es definido como la fase sólida del desecho y el filtrado como la fase líquida[30].
-- Determinar el peso de la fase líquida restando el peso del erlenmeyer vacío, del peso del erlenmeyer lleno con el filtrado después de la filtración.
-- El filtrado puede ser analizado o almacenado a 4 ºC hasta el momento del análisis.
-- Si el desecho contiene < 0,5% de sólidos secos, el filtrado se define como el “Extracto SPLP”. Si el desecho contiene > 0,5% de sólidos secos, se debe almacenar o analizar la fase líquida a y la fase sólida se le debe realizar el proceso de extracción después del de reducción de tamaño (si lo requiere acorde a las pruebas preliminares).
VI-ii.1.2 Reducción de tamaño fase sólida
Este proceso se aplica si la fase sólida requiere reducción de tamaño con base en las pruebas preliminares. Toda la fase sólida debe ser preparada machacando, cortando, moliendo hasta lograr las condiciones de tamaño de partícula.
VI-ii.1.3 Extracción de la fase sólida
Para desechos con > 0,5% de sólidos secos, y que no requieran reducción de tamaño de sólidos:
-- Transferir cuantitativamente la fase sólida al contenedor de extracción junto con el filtro usado en la separación líquido sólido.
Para desechos con > 0.5% de sólidos secos que requieren reducción de tamaño de partículas:
-- Transferir cuantitativamente la fase sólida después de la reducción de tamaño al contenedor de extracción junto con el filtro usado en la separación líquido sólido.
-- Determinar la cantidad del fluido de extracción requerido para la extracción por la siguiente fórmula:
![]()
Donde:
%S = porcentaje de sólidos determinado en el ensayo preliminar 1 Determinación preliminar del porcentaje de sólidos.
Wf = peso del desecho filtrado inicial o peso de la muestra total para 100% sólidos.
-- Lentamente adicionar esta cantidad al contenedor de extracción, cerrar de forma ajustada (se recomienda usar tapas de teflón para asegurar el sello) y asegurar en el mecanismo de agitación rotatorio.
-- Se debe realizar el proceso de extracción con un blanco como mínimo para cada 20 extracciones que se realicen en el recipiente de extracción.
-- Rotar a 30 ± 2 rpm por 18 ± 2 horas, la temperatura del laboratorio donde se realiza la extracción debe mantenerse a 23 ± 2 ºC durante este período[31].
-- Después de las 18 ± 2 horas de extracción, separar el material dentro del recipiente de extracción en sus componentes fases líquida y sólida por filtración a través de un nuevo filtro de fibra de vidrio (en este caso por bomba de vacío). En esta filtración final el papel de fibra de vidrio puede ser cambiado, si se requiere para facilitar la filtración. Los filtros deben ser enjuagados con ácido si se va a evaluar la movilidad de los metales.
VI-ii.1.4 Extracto SPLP
-- Si el desecho no contenía fase líquida inicial, el líquido filtrado después de 1.3 es el “Extracto SPLP”.
-- Si el desecho contiene fase líquida y sólidos <0,5% el filtrado obtenido en 1.1 Filtración es el “Extracto SPLP”
-- Si el desecho contiene fase líquida y sólidos > 0,5%, verificar si el líquido obtenido en 1.1 Filtración “fase líquida” y el filtrado en 1.3. Extracción de la fase sólida son compatibles, combínelos y este el “Extracto SPLP”. Si no lo son registrar el volumen de cada uno y analizarlos por separado. Ambos son “Extracto SPLP” y al final combinar matemáticamente sus resultados:
Donde:
V1 | Volumen primera fase |
| C1 | Concentración primera fase |
| V2 | Volumen segunda fase |
| C2 | Concentración segunda fase. |
-- Registrar el pH del Extracto inmediatamente después de su colección, tomar alícuotas para los diferentes análisis a realizar y preservar para su posterior análisis.
-- Las alícuotas para metales deben ser acidificadas con HNO3 a pH < 2. (si se observa precipitación tomar otra alícuota para análisis de metales, no acidificar, refrigerar a 4 ºC y analizar lo antes posible). Estas alícuotas deben ser digeridas en ácido excepto en aquellas circunstancias donde la digestión causa pérdidas de analitos metálicos. Si un análisis de un extracto sin digerir muestra que la concentración del analito metálico regulado excede el nivel regulatorio, el desecho se clasifica como peligroso y no se requiere digestión del extracto. Sin embargo los datos de extractos sin digerir no pueden ser usados para demostrar que el desecho no es peligroso.
-- Las otras alícuotas deben ser almacenadas bajo refrigeración 4 ºC hasta su análisis.
-- El extracto SPLP debe ser preparado y analizado acorde al método analítico apropiado.
VI-ii.2 Procedimiento para extracción de compuestos volátiles
Para obtener el extracto SPLP para compuestos volátiles solo se debe usar el dispositivo ZHE. El extracto obtenido del uso de este equipo no debe ser usado para evaluar la movilidad de analitos no volátiles (p.e. metales, pesticidas, etc.).
El equipo ZHE tiene aproximadamente 500 ml de capacidad interna. De esta forma puede acomodar como máximo 25 gramos de sólidos (definidos como la fracción de muestra para la cual no se puede forzar fuera líquido adicional al aplicar una presión de 50 PSI), debido a la necesidad de adicionar una cantidad de fluido de extracción igual a 20 veces el peso de la fase sólida.
Recomendaciones
-- Cargar el ZHE con la muestra solo una vez y no abrir el equipo hasta que se haya recolectado el extracto final. No se permite el rellenar el ZHE para obtener 25 gramos de sólidos. No permitir que el desecho, la fase líquida inicial o el extracto estén expuestos a la atmósfera por más del tiempo que sea absolutamente necesario. Cualquier manipulación de estos materiales debe ser hecha cuando estén fríos 4 ºC para minimizar las pérdidas de volátiles.
-- Si el desecho es 100% sólido pesar una submuestra de 25 gramos como máximo del desecho, registrar el peso y proceder con la preparación del equipo y materiales, en este caso no se requiere del paso de filtración de la fase líquida inicial.
-- Si el desecho contiene < 0,5% de sólidos secos la porción líquida del desecho, después de la filtración, se define como el extracto SPLP. Se debe filtrar bastante muestra de forma que la cantidad de líquido filtrado sea suficiente para todos los análisis de compuestos volátiles requeridos.
-- Para desechos conteniendo = 0,5% de sólidos secos, usar la información del porcentaje de sólidos para determinar la cantidad de muestra óptima para cargar dentro del ZHE. Los tamaños de muestra recomendados son los siguientes:
– Para desechos conteniendo < 5% de sólidos, pesar 500 gramos de submuestra y registrar el peso.
– Para desechos conteniendo = 5% de sólidos, determinar la cantidad de desecho a cargar dentro del ZHE como sigue:
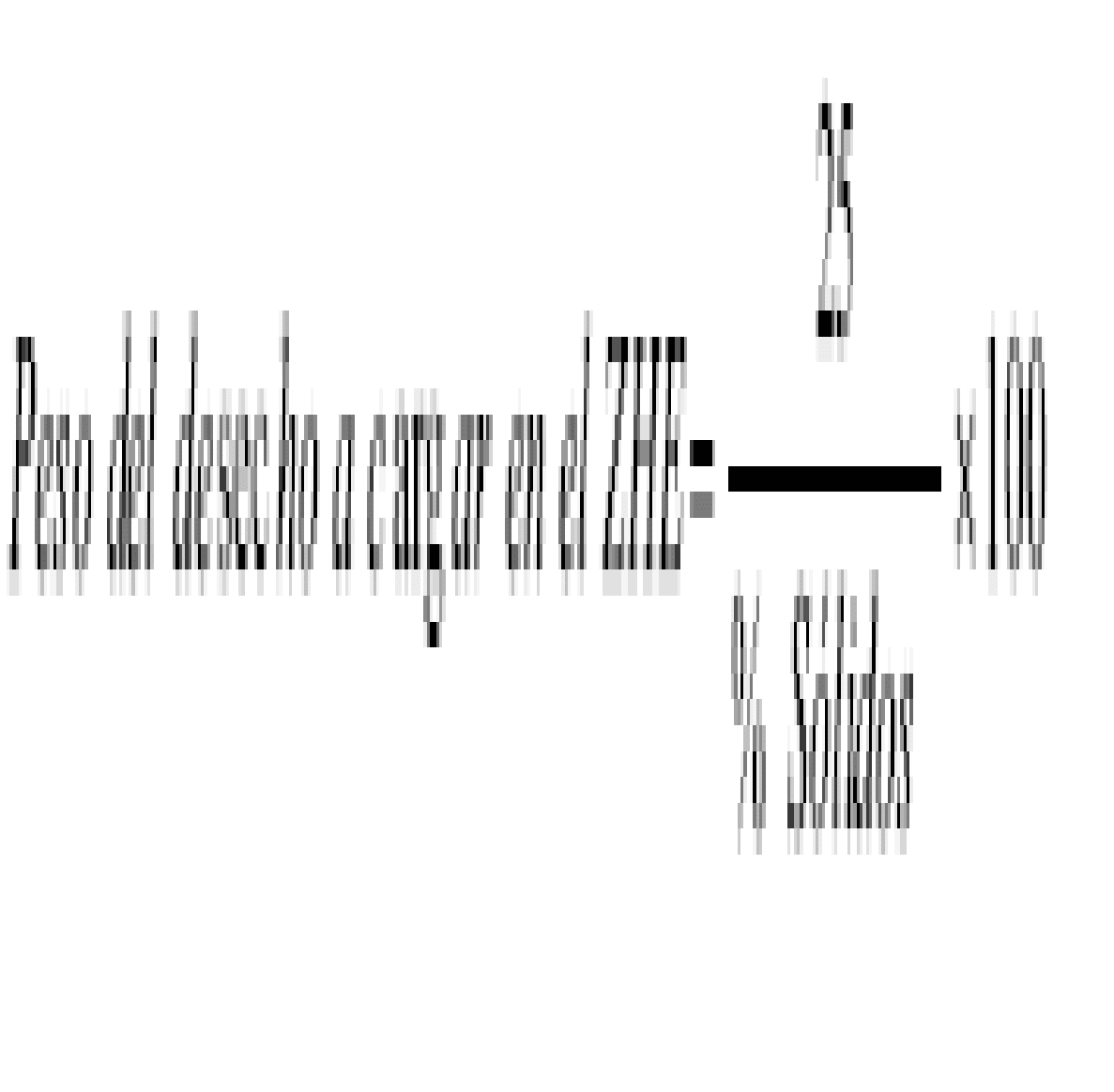
Pesar la submuestra del desecho del tamaño apropiado y registrar el peso.
-- Si se requiere reducción de tamaño de partícula de la porción sólida del desecho, preparar el desecho para la extracción machacando, cortando o moliendo la porción sólida del desecho a un área superficial o tamaño de partícula requerido. El desecho y el equipo de reducción apropiado deben ser refrigerados si es posible a 4 ºC antes de la reducción de tamaño de partícula. El equipo o medio usado para reducir el tamaño de las partículas no debe generar calor interna o externa. Si se requiere reducción de la fase sólida, la exposición del desecho a la atmósfera debe ser evitada en la medida de lo posible[32].
-- Para desechos lodosos no es necesario permitir la estanqueidad para que la fase sólida se sedimente. No se debe centrifugar los desechos antes de la filtración.
-- Se debe analizar como mínimo de un blanco usando el fluido de extracción No 1 cada 20 extracciones que se realicen.
VI-ii.2.1 Preparación del Equipo y Materiales del ZHE
-- Prepesar el contenedor de recolección del filtrado (el cual puede ser bolsa Tedlar® o jeringa dependiendo de las características del desecho como se mencionó en el numeral IV (Equipos y Materiales).
-- Colocar el pistón del ZHE dentro del cuerpo del equipo (puede ser de ayuda primero humedecer el empaque del pistón con el fluido de extracción). Ajustar el pistón dentro del cuerpo del ZHE hasta una altura en la cual se minimice la distancia que el pistón tenga que moverse una vez el ZHE esté cargado con la muestra (basado sobre los requerimientos de tamaño de la muestra). Asegurar el flanche de entrada/salida del gas (fondo del flanche) sobre el cuerpo del ZHE de acuerdo con las instrucciones del fabricante. Asegurar el filtro fibra de vidrio entre las pantallas de soporte y el lado. Fijar el flanche de entrada/salida del líquido (arriba de la pestaña) a un lado.
-- Pesar la cantidad de muestra requerida para el análisis de acuerdo a las recomendaciones anteriores.
-- Transferir cuantitativamente la muestra completa (fase líquida y sólida) lentamente al ZHE. Asegurar todos los accesorios y colocar el mecanismo en posición vertical (flanche de entrada/salida del gas en el fondo). No atar el dispositivo de recolección del extracto al plato superior. Si el material de desecho (>1% del peso de la muestra original) está adherido al contenedor usado para transferir la muestra al ZHE, determinar el peso de este residuo y restarlo del peso de la muestra para determinar el peso del desecho que será filtrado.
-- Conectar la línea de gas de la válvula de entrada/salida de gas (flanche inferior) y con la válvula de entrada/salida del líquido abierta, comenzar a aplicar lentamente presión de 1 a 10 PSI (o mas si es necesario) para forzar todo el headspace lentamente fuera del mecanismo de ZHE bajo una cabina. Con la primera aparición de líquido de la válvula de entrada/salida de líquido, rápidamente cerrar la válvula y dejar de aplicar presión. Si la filtración del desecho a 4 ºC reduce la cantidad de líquido comparado con la cantidad de líquido a temperatura de cuarto, se debe permitir que la muestra se caliente hasta temperatura de cuarto en el equipo antes de la filtración. Si el desecho es 100% sólido, lentamente incrementar la presión hasta un máximo de 50 PSI para forzar más del headspace fuera del equipo.
VI-ii.2.2 Filtración fase líquida inicial
-- Atar el dispositivo de recolección del filtrado prepesado a la válvula de entrada/salida de líquido y abrir la válvula. Comenzar a aplicar lentamente presión de 1 a 10 PSI para forzar a la fase líquida de la muestra dentro del dispositivo. Si no se observa líquido adicional pasar a través del filtro en un período de 2 minutos, lentamente incrementar la presión en incrementos de 10 PSI hasta un máximo de 50 PSI. Después de cada incremento de 10 PSI, si no se observa líquido adicional pasar a través del filtro en un período de 2 minutos, proceder al próximo incremento de 10 PSI. Cuando el flujo del líquido ha cesado tal que la filtración a 50 PSI no resulta en filtrado adicional en 2 minutos se debe parar la filtración. Cerrar la válvula de entrada/salida del líquido, descargar la presión del pistón y desconectar y pesar el dispositivo de recolección del filtrado[33].
-- El material en el ZHE se define como la fase sólida del desecho y el filtrado es definido como la fase líquida. La fase líquida puede ser analizada inmediatamente o almacenada a 4 ºC bajo condiciones de mínimo headspace hasta el momento de análisis[34].
VI-ii. 2.3 Determinación de la Cantidad de Fluido de Extracción
-- Determinar el peso del fluido de extracción No 3 para adicionar al equipo ZHE de la siguiente forma:
VI-ii.2.4 Llenado del equipo con el fluido de extracción
-- Con el ZHE en posición vertical, unir una línea del contenedor del fluido de extracción a la válvula de entrada/salida de líquido. La línea usada debe contener fluido de extracción fresco y debe ser prenivelado con el fluido para eliminar cualquier burbuja de aire en la línea. Liberar la presión de gas sobre el pistón del ZHE (desde la válvula de entrada/salida del gas), abrir la válvula de entrada/salida del líquido y comenzar la transferencia del fluido de extracción (por bombeo o un medio similar) dentro del ZHE. Continuar con el bombeo hasta que la cantidad apropiada del fluido sea introducida.
-- Inmediatamente después de que ha sido adicionado el fluido de extracción cerrar la válvula de entrada/salida de líquido y desconectar la línea del fluido de extracción. Verificar el ZHE para asegurar que todas las válvulas están en sus posiciones de cerrado. Manualmente rotar el equipo con 2 o 3 veces. Reposicionar el ZHE en la posición vertical con la válvula de entrada/salida del líquido hacia arriba. Presurizar el ZHE a 5 – 10 PSI (si es necesario) y lentamente abrir la válvula de entrada/salida del líquido para dejar salir cualquier headspace (dentro de una cabina) que pudo haberse introducido debido a la adición del fluido de extracción. Esta abertura debe hacerse rápidamente y debe parar con la primera aparición de líquido en la válvula. Re-presurizar el ZHE de 5 a 10 PSI y verificar que todos los accesorios del ZHE para asegurar que están cerrados.
VI-ii.2.5 Proceso de extracción
-- Localizar el ZHE en el aparato de agitación rotatorio y rotar a 30 ± 2 rpm por 18 ± 2 horas. La temperatura ambiente (p.e. temperatura del cuarto en el cual se realiza la extracción) debe mantenerse a 23 ± 2 ºC durante la agitación.
-- Finalizado el período de agitación de 18 ± 2 horas, verificar la presión dentro del pistón del ZHE rápidamente abriendo y cerrando la válvula de entrada/salida del gas y notar el escape de gas. Si la presión no se ha mantenido (p.e. no se observa liberación de gas), el equipo presenta una fuga. Se debe realizar el proceso de verificación de fugas presentado en el numeral de Equipos y Materiales, y realizar el proceso de extracción de nuevo con una nueva muestra del desecho. Si la presión dentro del equipo se ha mantenido, el material en el recipiente de extracción es separado en sus fases líquida y sólida.
VI-ii.2.6 Filtración del extracto
-- Filtrar a través del filtro de fibra de vidrio, usando el mismo proceso de filtración discutido anteriormente para ZHE de la fase líquida inicial. Si el desecho contenía una fase líquida inicial, el líquido puede ser filtrado directamente dentro del mismo dispositivo de recolección de filtrado que contenía la fase líquida inicial del desecho. Si la combinación de fluidos puede crear múltiples fases debe usarse un dispositivo de recolección de filtrado separado, o si no hay suficiente volumen dentro del dispositivo de recolección de filtrado.
-- Todo el extracto debe ser filtrado y recolectado en los casos en los cuales se usa bolsa Tedlar®, si el extracto es de múltiples fases, o si el desecho contenía una fase líquida inicial.
VI-ii.3 Extractos SPLP
-- Si el desecho original no contenía una fase líquida inicial el líquido filtrado del extracto es definido como el Extracto SPLP.
-- Si el desecho contenía una fase líquida inicial, el material líquido filtrado obtenido después del proceso de extracción y la fase líquida inicial se definen colectivamente como el Extracto SPLP.
-- Si el desecho no contenía sólidos o los sólidos secos eran <0,5 % la fase líquida inicial corresponde al Extracto SPLP.
Preparación de extracto para análisis.
-- Inmediatamente después de la recolección del extracto SPLP, preparar el extracto para análisis y almacenar con el mínimo espacio de cabeza a 4 ºC hasta su análisis. Se debe realizar el análisis del Extracto SPLP de acuerdo al método analítico apropiado (Ver Anexo 5).
-- Si las fases individuales van a ser analizadas por separado (p.e. que no sean miscibles), determinar el volumen de las fases individuales (a 0,5 %), realizar los análisis por separado y combinar los resultados matemáticamente usando una promedio ponderado por volumen.
Donde:
| V1 | Volumen primera fase (L) |
| C1 | Concentración del analito en la primera fase (mg/l) |
| V2 | Volumen segunda fase (L) |
| C2 | Concentración del analito en la segunda fase (mg/l). |
VII. RESULTADOS
Este método presenta el proceso de obtención del Extracto SPLP; obtenido este se deben realizar las determinaciones para cada analito de interés siguiendo los métodos analíticos adecuados (Anexo 5). Comparar las concentraciones de los analitos en el extracto TCLP con los niveles identificados en la regulación apropiada.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 6.3. Diagrama de Flujo – Evaluaciones preliminares.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 6.4. Diagrama de flujo – Ensayo para compuestos no-volátiles.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 6.5. Diagrama de flujo – Ensayo para compuestos volátiles.
VIII. ASEGURAMIENTO DE LA CALIDAD
-- Se debe analizar como mínimo un blanco (usando el mismo fluido de extracción usado para las muestras) cada 20 extracciones realizadas.
-- Se debe realizar un ensayo con matriz dopada para cada tipo de desecho (p.e. lodos de plantas de tratamiento de aguas residuales, suelos contaminados, etc.) a menos que los resultados excedan el nivel regulatorio y los datos estén siendo usados únicamente para demostrar que el desecho excede el nivel regulatorio. Se requiere que como mínimo una matriz dopada sea analizada para cada bache de análisis. Como mínimo, siga la guía de adición para matriz estándar de cada método analítico.
– Los dopajes a la matriz se adicionan después de la filtración del Extracto SPLP y antes de la preservación. Los dopajes no deben ser adicionados antes de la extracción de la muestra.
– En la mayoría de los casos, los dopajes deben ser adicionados a una concentración equivalente al nivel regulatorio correspondiente. Si la concentración del analito es menor que la mitad del nivel regulatorio, la concentración de dopaje puede ser tan bajo como la mitad de la concentración del analito, pero no debe ser menor que cinco veces el límite de detección del método. Para evitar diferencias en el efecto matriz, los dopajes deben ser adicionados al mismo volumen nominal del Extracto SPLP del que fue analizado en la matriz sin dopar.
– El propósito del dopaje es monitorear el comportamiento del método analítico usado, y determinar si existen interferencias de matriz. Cuando la recuperación de la matriz dopada es por debajo del comportamiento del método analítico esperado, se deben usar otros métodos de calibración interna, modificaciones de los métodos analíticos, o uso de métodos analíticos alternativos de forma que se alcance una medición confiable de la concentración del analito.
– El porcentaje de recuperación de matrices dopadas son calculados por la siguiente fórmula:
Donde:
| Xs | valor medido para la muestra dopada |
| Xu | valor medido para la muestra sin dopaje |
| K | valor conocido de dopaje en la muestra. |
-- Todos las mediciones de control de calidad descritos en los métodos analíticos apropiados se deben seguir.
-- El uso de métodos de cuantificación de calibración interna deben ser empleados para contaminantes metálicos si, a) la recuperación del contaminante desde el extracto TCLP es menor de 50% y la concentración no excede el nivel regulatorio o, b) la concentración del contamínate medido en el extracto esta dentro del 20% del nivel regulatorio apropiado.
– El método de adiciones estándares debe ser empleado como un método de cuantificación de calibración interna para cada uno de los contaminantes metálicos.
– El método de adiciones estándares requiere la preparación de estándares de calibración en la matriz de la muestra en vez de prepararlos en agua grado reactivo o blanco de la solución. Se requiere tomar 4 alícuotas idénticas de la solución y adicionar cantidades conocidas de estándar a 3 de estas alícuotas. La cuarta alícuota es la desconocida. Preferiblemente, la primera adición debe ser preparada de forma tal que la concentración resultante sea aproximadamente el 50% de la concentración esperada de la muestra. La segunda y tercera adición deben ser preparadas de forma tal que las concentraciones sean aproximadamente el 100% y el 150% de la concentración esperada de la muestra. Las cuatro alícuotas son llevadas al mismo volumen final por adición de agua grado reactivo o una solución de blanco, y pueden necesitar ajuste de dilución para mantener la señal en el rango lineal de la técnica del instrumento. Se analizan las cuatro alícuotas.
– Graficar la señal del instrumento o concentración derivada de la calibración externa como la variable dependiente (eje y) versus la concentración de las adiciones de estándar como la variable independiente (eje x). Determinar el intercepto de la abcisa (variable independiente, eje x) la cual será la concentración desconocida.
– Alternativamente, restar la señal del instrumento o la concentración derivada de la calibración externa de la matriz desconocida de la señal o concentración derivada de la calibración externa de las adiciones estándares. Graficar o hacer una regresión lineal de la señal del instrumento corregido o de la concentración derivada de la calibración externa corregida como variable dependiente versus la variable independiente. Calculara las concentraciones para las concentraciones desconocidas en la curva de calibración interna como si fuera una curva de calibración externa.
-- La siguiente tabla presenta los períodos de tiempo de mantenimiento máximo de la muestra entre las diferentes etapas del procesamiento para el análisis de TCLP
Tiempo máximo de mantenimiento de muestra (días)
| Desde | Toma de muestra | Extracción | Preparación del extracto | Tiempo total gastado |
| Hasta | Extracción | Preparación del extracto | Análisis | |
| Compuestos volátiles | 14 | NA | 14 | 28 |
| Compuestos semivolátiles | 14 | 7 | 40 | 61 |
| Mercurio | 28 | NA | 28 | 56 |
| Metales (Excepto mercurio) | 180 | NA | 180 | 360 |
NA = No aplica
Si los tiempos de mantenimiento son excedidos, los valores obtenidos pueden ser considerados como concentraciones mínimas. Si se excede el tiempo de mantenimiento no es aceptable para establecer si el desecho no excede el nivel regulatorio. Si se excede el tiempo de mantenimiento no invalida la caracterización del desecho si este excede el nivel regulatorio.
IX. INFORMACION ESTADISTICA
IX-i. Precisión
Compuestos orgánicos semivolátiles y metales
En la siguiente tabla se presenta el resultados (EPA) de un suelo el cuales fue dopado con el fluido No 1. La concentración de los contaminantes lixiviados de los suelos fueron reproducibles dado por la moderada desviación estándar relativa (%SD) de las recuperaciones (en promedio 29% para los compuestos y elementos analizados).
| Compuesto | Cantidad dopada (µg) | Cantidad recuperada (µg)* | % RSD |
Semivolátiles
| Bis(2-cloroetil)-eter | 1040 | 834 | 12,5 |
| 2-clorofenol | 1620 | 1010 | 6,8 |
| 1,4 -diclorobenceno | 2000 | 344 | 12,3 |
| 1,2-diclorobenceno | 8920 | 1010 | 8,0 |
| 2-metilfenol | 3940 | 1860 | 7,7 |
| Nitrobenceno | 1010 | 812 | 10,0 |
| 2,4-dimetilfenol | 1460 | 200 | 18,4 |
| Hexaclorobutadieno | 6300 | 95 | 12,9 |
| Acenafteno | 3640 | 210 | 8,1 |
| 2,4 –dinitrofenol | 1300 | 896** | 6,1 |
| 2,4-dinitrotolueno | 1900 | 1150 | 5,4 |
| Hexaclorobenceno | 1840 | 3,7 | 12,0 |
| Gamma BHC (Lindano) | 7440 | 230 | 16,3 |
| Beta BHC | 640 | 35 | 13,3 |
Metales
| Plomo | 5000 | 70 | 4,3 |
| Cadmio | 1000 | 387 | 2,3 |
*Análisis triplicados.
** Análisis duplicados, un valor fue rechazado como un valor extremo con un 90% de nivel de confianza usando el ensayo Q Dixon.
Compuestos orgánicos volátiles
Cuatro suelos diferentes fueron dopados y analizados. Los resultados se presentan en la siguiente tabla. Los suelos 1 y 2 eran suelos contaminados. Los suelos 3 y 4 eran mezclas de suelo con bajo contenido orgánico y dos diferentes lodos municipales. Las replicas de lixiviado de los suelos 3 y 4 presentaron menor precisión que los lixiviados de suelos contaminados.
| Suelo 1 | Suelo 2 | Suelo 3 | Suelo 4 | |||||
| Compuesto | %R | %RSD | %R | %RSD | %R | %RSD | %R | %RSD |
| Acetona | 44 | 12.4 | 43.8 | 2.25 | 116.0 | 11.5 | 21.3 | 71.4 |
| Acrilonitrilo | 52.5 | 68.4 | 50.5 | 70.0 | 49.3 | 44.9 | 51.8 | 4.6 |
| Benceno | 47.8 | 8.29 | 34.8 | 16.3 | 49.8 | 36.7 | 33.4 | 41.1 |
| 1-butanol | 55.5 | 2.91 | 49.2 | 14.6 | 65.5 | 37.2 | 73.0 | 13.9 |
| Disulfuro de carbono | 21.4 | 16.4 | 12.9 | 49.5 | 36.5 | 51.5 | 21.3 | 31.5 |
| Tetracloruro de carbono | 40.6 | 18.6 | 22.3 | 29.1 | 36.2 | 41.4 | 24.0 | 34.0 |
| Clorobenceno | 64.4 | 6.76 | 41.5 | 13.1 | 44.2 | 32.0 | 33.0 | 24.9 |
| Cloroformo | 61.3 | 8.04 | 54.8 | 16.4 | 61.8 | 29.1 | 45.8 | 38.6 |
| 1,2 -dicloroetano | 73.4 | 4.59 | 68.7 | 11.3 | 58.3 | 33.3 | 41.2 | 37.8 |
| 1,1-dicloroeteno | 31.4 | 14.5 | 22.9 | 39.3 | 32.0 | 54.4 | 16.8 | 26.4 |
| Acetato de etilo | 76.4 | 9.65 | 75.4 | 4.02 | 23.0 | 119.8 | 11.0 | 115.5 |
| Etilbenceno | 56.2 | 9.22 | 23.2 | 11.5 | 37.5 | 36.1 | 27.2 | 28.6 |
| Etileter | 48.0 | 16.4 | 55.1 | 9.72 | 37.3 | 31.2 | 42.0 | 17.6 |
| Isobutanol | 0.0 | ND | 0.0 | ND | 61.8 | 37.7 | 76.0 | 12.2 |
| Cloruro de metileno | 47.5 | 30.3 | 42.2 | 42.9 | 52.0 | 37.4 | 37.3 | 16.6 |
| 2-butanona | 56.7 | 5.94 | 61.9 | 3.94 | 73.7 | 31.3 | 40.6 | 39.0 |
| Metil isobutil cetona | 81.1 | 10.3 | 88.9 | 2.99 | 58.3 | 32.6 | 39.8 | 40.3 |
| 1,1,1,2 -tetracloroetano | 69.0 | 6.73 | 41.1 | 11.3 | 50.8 | 31.5 | 36.8 | 23.8 |
| 1,1,2,2 -tetracloroetano | 85.3 | 7.04 | 58.9 | 4.15 | 64.0 | 25.7 | 53.6 | 15.8 |
| Tetracloroeteno | 45.1 | 12.7 | 15.2 | 17.4 | 26.2 | 44.0 | 18.6 | 24.2 |
| Tolueno | 59.2 | 8.06 | 49.3 | 10.5 | 45.7 | 35.2 | 31.4 | 37.2 |
| 1,1,1 -tricloroetano | 47.2 | 16.0 | 33.8 | 22.8 | 40.7 | 40.6 | 26.2 | 38.8 |
| 1,1,2 - tricloroeteno | 76.2 | 5.72 | 67.3 | 8.43 | 61.7 | 28.0 | 46.4 | 25.4 |
| Tricloroeteno | 54.5 | 11.1 | 39.4 | 19.5 | 38.8 | 40.9 | 25.6 | 34.1 |
| Triclorofluorometano | 20.7 | 24.5 | 12.6 | 60.1 | 28.5 | 34.0 | 19.8 | 33.9 |
| 1,1,2 - triclorotrifluoroetano | 18.1 | 26.7 | 6.95 | 58.0 | 21.5 | 67.8 | 15.3 | 24.8 |
| Cloruro de vinilo | 10.2 | 20.3 | 7.17 | 72.8 | 25.0 | 61.0 | 11.8 | 25.4 |
%R = Porcentaje de recuperación.
X. REFERENCIAS
Método 1311. Procedimiento de lixiviación para la característica de toxicidad. SW 846. Revisión 0 julio 1992
6.3 Toxicidad aguda para daphnia
Este método corresponde al método de Toxicidad aguda para Daphnia ensayo extremo (una traducción ajustada del método C2 Acute Toxicity for Daphnia de la Comunidad Europea), evaluado en la fracción adaptada de agua (WAF-por sus siglas en inglés) de un desecho.
I. ALCANCE Y APLICABILIDAD
Este método junto con el ensayo de inhibición de crecimiento de algas permite clasificar un desecho complejo como desecho peligroso por toxicidad acuática. Los desechos complejos son aquellos que contienen sustancias para las cuales no hay datos de toxicidad acuática o donde el desecho es una mezcla sin caracterizar.
El ensayo consiste en determinar el porcentaje de inmovilización de Daphnia con 100% de la fracción adaptada de agua (WAF) del desecho contra un control. Si el porcentaje de inmovilización es = 50% el desecho debe ser clasificado como ecotóxico.
En aquellos casos cuando el resultado de toxicidad aguda en Daphnia e Inhibición de algas están cercanos al límite del 50% es necesario hacer evaluación adicional con peces.
II. PRINCIPIO DEL METODO
El desecho es preparado adicionando agua en una relación especificada, mezclando por un tiempo determinado y después de un período de quietud es obtenida la fracción ajustada de agua (WAF) la cual es una fracción acuosa que contiene la fracción disuelta y/o suspendida y/o emulsificada del desecho.
Daphnia son expuestas a 100% WAF del desecho por 48 horas. Si se usa un ensayo más corto, se debe presentar la justificación en el informe. Al final del período se evalúa la inmovilización de Daphnia comparado con el control y se calcula el porcentaje de inmovilización.
Se usa un sistema estático para este método, por lo cual las soluciones de ensayo no son renovadas durante el período de exposición.
III. INTERFERENCIAS
No hay interferencias reportadas para este método.
IV. EQUIPOS Y MATERIALES
Se deben usar equipos y aparatos normales de laboratorio. Los equipos que estarán en contacto con las soluciones a evaluar deben ser preferiblemente de vidrio completamente.
-- Medidor de oxígeno (con micro electrodo u otro equipo adecuado para medir oxígeno disuelto en muestras con bajo volumen).
-- Aparato adecuado para control de temperatura.
-- pHmetro
-- Equipo para la determinación de dureza del agua.
V. REACTIVOS
-- Fracción ajustada de agua del desecho (WAF).
Al desecho se le adiciona agua en una relación de 100 mg de desecho por un litro de agua, y se mezcla con este, colocándolo en un vortex del agua creado por un agitador magnético en aspiración (la agitación debe ser suficientemente vigorosa para crear un vortex), el período de mezcla para desechos complejos conteniendo metales es de 7 días y para otros tipos de desechos es de 48 horas.
Después del período de mezcla se permite la sedimentación por una hora después de la cual se retira el WAF del aspirador. Se descarta el desecho sin disolver y sin dispersar. El ensayo debe ser realizado sin ajuste de pH. Si hay evidencia de cambios marcados en el pH, el ensayo debe repetirse con ajuste de pH y debe ser reportado en los resultados.
-- Agua de ensayo
Agua reconstituida (Ver Anexo 6 e ISO 6341-1989 “Determination of inhibition of mobility of Daphnia Magna Straus”). Para evitar la necesidad de aclimatación antes del ensayo, es recomendado que el agua de cultivo presente una calidad similar (pH, dureza) al agua usada en el ensayo.
-- Sustancia de referencia
Se puede evaluar una sustancia de referencia como un medio para demostrar que bajo las condiciones del ensayo en el laboratorio la sensibilidad de las especies de ensayo no han cambiado significativamente. En el Anexo 7, se presentan los resultados del ensayo para cuatro sustancias diferentes.
-- Organismos de ensayo
Daphnia magna es la especie de ensayo preferida aunque también se permite Daphnia pulex. Los organismos del ensayo deben tener menos de 24 horas al inicio del ensayo, sepas de laboratorio, libres de enfermedades y con un historial conocido (p.e. reproducción – cualquier pretratamiento, etc.).
VI. PROCEDIMIENTO
Se debe realizar un control sin la sustancia que se está evaluando.
-- Daphnia son expuestas al WAF de acuerdo a la siguiente descripción.
– Duración: preferiblemente 48 horas.
– Número de animales: al menos 20 en cada una de las concentraciones de ensayo preferiblemente divididas en cuatro baches de 5 animales cada uno o dos baches de 10 animales.
– Carga: 100% WAF.
– Concentración del ensayo: 100% WAF.
– Agua de ensayo: para el control.
– Luz: un ciclo de luz-oscuridad es opcional.
– Temperatura: La temperatura del ensayo debe estar entre los 18 y 22 ºC, pero para cada ensayo individual debe ser constante dentro de ± 1 ºC.
– Aireación: Las soluciones a evaluar no deben ser aireadas.
– Alimentación: ninguna.
El pH y la concentración de oxígeno del control y del WAF deben ser medidos al final del ensayo, el pH de las soluciones a evaluar no deben ser modificadas. Compuestos volátiles deben ser evaluados en contenedores cerrados completamente llenos, con bastante cantidad para prevenir la falta de oxígeno.
Daphnia son inspeccionadas al menos cada 24 horas de exposición y de nuevo después de 48 horas.
VII. RESULTADOS
Para cada período de observaciones (24 y 48 horas), calcular el porcentaje de inmovilización promedio contra el control. Si se observa que la estabilidad o homogeneidad de la sustancia de ensayo no puede ser mantenido, se debe reportar y tener esto en consideración en la interpretación de los resultados.
El reporte debe incluir la siguiente información:
Información acerca del organismo de ensayo (nombre científico, variedad, proveedor o fuente, pre-tratamiento, método de reproducción-incluyendo fuente, clase y cantidad de alimento, frecuencia de alimentación).
Fuente del agua de dilución y principales características químicas (p.e. pH, temperatura, dureza).
Porcentaje de inmovilización para 100% WAF.
Descripción de los equipos del ensayo.
Régimen de luz.
Concentraciones de oxigeno disuelto, pH y temperatura de las soluciones de ensayo.
Evidencia de que los criterios de calidad han sido realizados.
Si se usa una sustancia de referencia, los resultados obtenidos.
VIII. ASEGURAMIENTO DE LA CALIDAD
La inmovilización en los controles no debe exceder el 10% al final del ensayo.
Daphnia en los grupos de control no deben estar atrapadas en la superficie del agua.
Es deseable que la concentración de oxígeno disuelto en los contenedores del ensayo permanezcan por encima de los 3 mg/l durante el ensayo. Sin embargo, en ninguna circunstancia debe disminuir la concentración de oxigeno disuelto por debajo de 2 mg/l.
La concentración de la sustancia de ensayo debe ser mantenida dentro del 80% de la concentración inicial durante la duración del ensayo.
El pH no debe variar en más de una unidad durante el ensayo.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 6.6. Diagrama de Flujo Toxicidad agua para Daphnia.
IX. INFORMACION ESTADISTICA
No se reporta.
X. REFERENCIAS
C2. Acute Toxicity for Daphnia. Página internet. Del Annex V Testing Methods
Guía Técnica WM2. Desechos peligrosos. Interpretación de la definición y clasificación de los desechos peligrosos. Segunda Edición. SEPA Scottish Environment Protection Agency, Environment and Heritage Service and Environment Agency. Página web. www.environment-agency.gov.uk.
OECD-Organización para la cooperación y desarrollo económico -Serie sobre Pruebas y Ensayos No 23. Documento Guía sobre ensayos de toxicidad acuática de sustancias y mezclas dificultosas. ENV/JM/MONO(2000)6.
6.4 Ensayo de inhibición de algas
Este método corresponde al ensayo de inhibición de algas con ensayo extremo (una traducción ajustada del método C3 Algal Inhibition Test de la Comunidad Europea), evaluado en la fracción ajustada de agua (WAF-por sus siglas en inglés) de un desecho.
I. ALCANCE Y APLICABILIDAD
Este método junto con el ensayo de inmovilización de Daphnia permite clasificar un desecho complejo como desecho peligroso por toxicidad acuática. Los desechos complejos son aquellos que contienen sustancias para las cuales no hay datos de toxicidad acuática o donde el desecho es una mezcla sin caracterizar.
El ensayo consiste en determinar el porcentaje de reducción de crecimiento y reducción de la tasa de crecimiento de una especie de alga verde unicelular con 100% de la fracción ajustada de agua (WAF) del desecho contra un control. Si los porcentajes de reducción son = 50% el desecho debe ser clasificado como ecotóxico. Este ensayo relativamente breve (72 horas) puede evaluar el efecto sobre varias generaciones
En aquellos casos cuando el resultado de toxicidad aguda en Daphnia e Inhibición de algas están cercanos al límite del 50% es necesario hacer evaluación adicional con peces.
II. PRINCIPIO DEL METODO
El desecho es preparado adicionando agua en una relación especificada, mezclando por un tiempo determinado y después de un período de quietud es obtenida la fracción acomodada de agua (WAF) la cual es una fracción acuosa, conteniendo la fracción disuelta y/o suspendida y/o emulsificada del desecho.
Cultivos de algas verdes seleccionadas de crecimiento exponencial son expuestas a 100% WAF por varias generaciones bajo condiciones definidas.
Las soluciones de ensayo son incubadas por un período de 72 horas, durante las cuales la densidad celula[35] r en cada solución es medida al menos cada 24 horas. Se determina la inhibición en el crecimiento en relación a un cultivo de control.
III. INTERFERENCIAS
No se reportan interferencias.
IV. EQUIPOS Y MATERIALES
Se deben usar equipos y aparatos normales de laboratorio.
Vidriería de volumen adecuado (p.e. recipiente cónico de 250 ml son apropiados cuando el volumen de la solución de ensayo es de 100 ml). Todos los recipientes deben ser idénticos con respecto a material y dimensiones.
Aparato de cultivo
Cámara o cabina en la cual una temperatura en el rango de 21 a 25 ºC pueda ser mantenida a ± 2 ºC, e iluminación uniforme y continua suministrada en un rango espectral de 400 a 700 nm. Si los cultivos de control han logrado las tasas de crecimiento recomendadas, se puede asumir que las condiciones de crecimiento, incluyendo la intensidad de la luz, han sido adecuadas.
Se recomienda usar para el nivel promedio de las soluciones de ensayo, una intensidad de luz en el rango de 60 a 120 µE.m-2.s-1 (35 a 70 x 1018 fotones.m-2.s-1) cuando se mide en el rango de 400 a 700 nm usando un receptor de luz apropiado. Para instrumentos de medición de luz calibrados en luxes, un rango equivalente de 6000 a 10000 1x es aceptable.
La intensidad de la luz puede ser obtenida usando cuatro a siete lámparas fluorescentes de 30W tipo blanco universal (temperatura de color de aproximadamente 4300 K) a una distancia de 0,35 m de los cultivos de algas.
Equipo de conteo directo de células vivas (p.e. un microscopio con cámara de conteo). Sin embargo, otros procedimientos (fotometría, turbidimetría) pueden ser usados si presentan sensibilidad suficiente y si muestran una buena correlación con la densidad celular.
V. REACTIVOS
Fracción ajustada de agua del desecho (WAF)
Al desecho se le adiciona agua en una relación de 100 mg de desecho por un litro de agua, y se mezcla con este, colocándolo en un vortex del agua creado por un agitador magnético en aspiración (la agitación debe ser suficientemente vigorosa para crear un vortex). El período de mezcla para desechos complejos conteniendo metales es de 7 días y para otros tipos de desechos es de 48 horas.
Después del período de mezcla se permite la sedimentación por una hora después de la cual se retira el WAF del aspirador. Se descarta el desecho sin disolver y sin dispersar.
El ensayo debe ser realizado sin ajuste de pH. Si hay evidencia de cambios marcados en el pH, el ensayo debe repetirse con ajuste de pH y debe ser reportado en los resultados. En aquellos casos, el valor de pH del WAF debe ser ajustado al valor de pH de la solución de agua a menos que existan razones específicas para no hacerlo. Es preferible usar para este propósito acido clorhídrico e hidróxido de sodio. Si se presenta cualquier tipo de reacción química o precipitación física del compuesto de ensayo por el ajuste de pH, se debe reportar en el informe.
Medio de ensayo
El agua debe ser agua destilada de buena calidad, o desionizada con una conductividad menor de 5 µS/cm. El equipo para la destilación del agua no debe contener ninguna parte de cobre.
Los siguientes medios son recomendados. Cuatro soluciones stock son preparadas, de acuerdo a la siguiente tabla. Las soluciones stock son esterilizadas por filtración de membrana o autoclavado, y almacenadas a la oscuridad a 4 ºC. La solución stock No 4 debe ser esterilizada solo por filtración por membrana. Estas soluciones stock son diluidas para lograr las concentraciones de nutrientes finales en la solución de ensayo.
| Nutriente | Concentración en la solución stock | Concentración final en la solución de ensayo |
| Solución Stock No 1: macronutrientes NH4Cl MgCl2.6H2O CaCl2.2H2O MgSO4.7H2O KH2PO4 | 1,5 g/L 1,2 g/L 1,8 g/L 1,5 g/L 0,16 g/L | 15 mg/l 12 mg/l 18 mg/l 15 mg/l 1,6 mg/l |
| Solución stock 2: Fe-EDTA FeCl3.6H2O Na2EDTA.2H2O | 80 mg/l 100 mg/l | 0,08 mg/l 0,1 mg/l |
| Solución Stock 3: elementos traza H3BO3 MnCl2.4H2O ZnCl2 CoCl2.6H2O CuCl2.2H2O Na2MoO4.2H2O | 185 mg/l 415 mg/l 3 mg/l 1,5 mg/l 0,01 mg/l 7 mg/l | 0,185 mg/l 0,415 mg/l 3 x 10-3 mg/l 1,5 x 10-3 mg/l 10-5 mg/l 7 x 10-3 mg/l |
| Solución Stock 4: NaHCO3 NaHCO3 | 50 g/L | 50 mg/l |
El pH del medio después de equilibrio con aire es aproximadamente 8.0.
Sustancia de Referencia
Se puede evaluar una sustancia de referencia como un medio para demostrar que bajo las condiciones de ensayo en el laboratorio la sensibilidad de las especies de ensayo no han cambiado significativamente. Si se usa una sustancia de referencia, se deben presentar los resultados en el informe.
Como sustancia de referencia se puede usar dicromato de potasio, pero su color puede afectar la intensidad y calidad de la luz disponible para las células y también en las determinaciones espectrofotométricas utilizadas. El dicromato de potasio ha sido utilizado como sustancia de prueba inter-laboratorio internacional (Ver Anexo 8).
Organismos del ensayo
Se sugiere el uso de especies de algas verdes de rápido crecimiento para el cultivo y el ensayo. Se prefieren las siguientes especies:
Selenastrum capricornutum, p.ej. ATCC [36] 22662 o CCAP[37] 278/4
Scenedesmus subspicatus, p.ej. 86.81 SAG[38]
Si se usan otras especies, se debe reportar la variedad.
VI. PROCEDIMIENTO
Las dos mediciones de crecimiento (biomasa y tasa de crecimiento) pueden resultar en mediciones muy dispersas de la inhibición del crecimiento.
Densidad celular inicial.
Se recomienda que la densidad celular inicial en los ensayos sea aproximadamente 10-4 células/ml para Selenastrum capricornutum y Scenedesmus subspicatus. Cuando se usan otras especies la biomasa debe ser comparable.
Concentraciones de la sustancia a evaluar.
La evaluación se realiza con 100% WAF.
Réplicas y controles.
El diseño del ensayo debe incluir tres réplicas para la evaluación del 100% WAF. Se debe evaluar tres controles sin sustancia.
Comportamiento del ensayo.
El cultivo de ensayo se prepara adicionando 100% WAF a un precultivos de algas (Ver Anexo 7).
Los recipientes de cultivo son agitados y ubicados en un aparato de cultivo. Las células de algas son guardadas en suspensión por agitación o burbujeo con aire, para mejorar el intercambio de gas y reducir la variación de pH en las soluciones. Los cultivos deben ser mantenidos a temperatura en el rango de 21 a 25 ºC, controlado a ± 2 ºC.
La densidad celular es determinada en cada recipiente como mínimo a las 24, 48 y 72 horas después del inicio del ensayo. El medio de algas filtrado conteniendo la concentración apropiada de la sustancia a evaluar es usado para determinar la concentración de fondo cuando se usan mediciones de densidad celular diferentes al método de conteo directo.
Medir el pH al inicio del ensayo y a las 72 horas. El pH de los controles no debe desviarse normalmente por más de 1,5 unidades durante el ensayo.
Evaluación de sustancias volátiles.
No hay datos de formas generalmente aceptadas para la evaluación de sustancias volátiles. Cuando se conoce que una sustancia presenta tendencia a ser vaporizada, se puede usar un recipiente cerrado con espacio de cabeza. La posibilidad de almacenar CO2 debe ser considerada cuando se calcula el espacio de cabeza de los recipientes cerrados. Se han propuesto variaciones a este método[39].
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 6.7. Diagrama de flujo Ensayo de inhibición de algas.
RESULTADOS
La medición de la densidad celular en el cultivo de ensayo de 100% WAF y los controles son tabulados para cada uno de los tiempos de medición. El valor medio de la densidad celular para 100% WAF y para los controles son calculados, y se calcula el porcentaje de reducción de crecimiento de biomasa y el porcentaje de reducción de la tasa de crecimiento de la forma explicada a continuación.
Comparación de áreas bajo la curva de crecimiento.
El área entre la curva de crecimiento y la linea horizontal N=No puede ser calculada de acuerdo a la fórmula:
| Donde: | |
| A | área, |
| N0 | número de células/ml en el tiempo t0 (comienzo del ensayo) |
| N1 | medición del número de células/ml a t1 |
| Nn | medición del número de células/ml en el tiempo n |
| t1 | tiempo de la primera medición después del comienzo del ensayo, |
| tn | tiempo de la nva medición después del inicio del ensayo |
| n | número de las mediciones tomadas después del inicio del ensayo. |
El porcentaje de inhibición del crecimiento celular para 100% WAF (IA) es calculada de acuerdo a la siguiente fórmula:
Donde:
Ac = área entre la curva de crecimiento del control y la línea horizontal N= No
At = área entre la curva de crecimiento para 100% WAF y la línea horizontal N = No
Comparación con las Tasas de Crecimiento.
El promedio de la tasa de crecimiento específica (µ) para cultivos de crecimiento exponencial pueden ser calculados con la siguiente fórmula
Donde:
t0 es el tiempo al inicio del ensayo.
Alternativamente, la tasa de crecimiento específica promedio puede ser derivada de la pendiente de la línea de regresión en el gráfico de ln N versus tiempo.
El porcentaje de inhibición de la tasa de crecimiento específica para 100% WAF (Ií t) se calcula a partir de la siguiente fórmula:
Donde:
íc = tasa de crecimiento específica del control
ít = tasa de crecimiento específica promedio para 100% WAF.
La tasa de crecimiento específica es un término logarítmico, y pequeños cambios en la tasa de crecimiento pueden generar grandes cambios en la biomasa. Por tal razón los valores de EbC y ErC no son comparables numéricamente.
El reporte debe incluir la información siguiente:
Sustancia a evaluar: datos de identificación.
Organismo de ensayo: origen, cultivo de laboratorio, variedad, método de cultivo
Condiciones del ensayo
– Fecha de inicio y terminación del ensayo y su duración
– Temperatura
– Composición del medio
– Aparato de cultivo
– pH de las soluciones al inicio y al final del ensayo (se debe suministrar una explicación si desviaciones mayores a 1,5 fueron observadas)
– Intensidad de la luz y calidad
Resultados
– Densidad celular en cada recipiente para cada punto de medición e indicar el método de medición de la densidad celular
– Valores promedios de la densidad celular
– Curvas de crecimiento
– Otros efectos observados
VII. ASEGURAMIENTO DE LA CALIDAD
La densidad celular en el cultivo de control debe incrementar en un factor de al menos 16 en tres días.
Las concentraciones de las sustancias de prueba deben ser mantenidas en un 80% de la concentración inicial a través del tiempo correspondiente a la duración del ensayo.
VIII. INFORMACION ESTADISTICA
No se reporta.
IX. REFERENCIAS
C3. Algal Inhibition Test del Annex V Testing Methods.
Guía Técnica WM2. Desechos peligrosos. Interpretación de la definición y clasificación de los desechos peligrosos. Segunda Edición. SEPA Scottish Environment Protection Agency, Environment and Heritage Service and Environment Agency. Página web. www.environment-agency.gov.uk.
OECD-Organización para la cooperación y desarrollo económico -Serie sobre Pruebas y Ensayos No 23. Documento Guía sobre ensayos de toxicidad acuática de sustancias y mezclas dificultosas. ENV/JM/MONO(2000)6
6.5 Diagrama metodológico determinación característica de toxicidad
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura 6.8. Arbol de decisión para ejecución de pruebas relativas a clasificación de residuos tóxicos.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Tabla 1
Guía para la selección de los diseños de muestreo
< TABLA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Tabla 2
Valores tabulados de “T” para la evaluación de desechos sólidos
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Tabla 3
Concentraciones estándares TCLP
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Tabla 4
Guía de selección del dispositivo. Medio y sitio de recolección de la muestra
< TABLA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Tabla 4
Guía de selección del dispositivo. Medio y sitio de recolección de la muestra
< TABLA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
TABLA 5
Descripción del medio enumerado en la Tabla 4
< TABLA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Tabla 6
Guía de selección del dispositivo. Factores específicos del dispositivo
< TABLA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Procedimiento para la aplicación de cada diseño de muestreo
< TABLA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Estrategias para el muestreo de desechos heterogéneos
IX.1 Introducción
“Desechos heterogéneos” incluyen estructuras, escombros, materiales de desecho de construcción, contenedores (barriles, tanques, y envases de pintura), desechos sólidos de laboratorios, procesos de manufactura, y desechos como componentes electrónicos, baterías, y partes de carros. Desechos heterogéneos pueden proponer cambios en el desarrollo e implementación del programa de diseño de muestreo debido a la variedad física en forma, tamaño y composición del material y la falta de herramientas y técnicas para muestrear desechos heterogéneos. La aplicación de técnicas de muestreo convencional en desechos heterogéneos es difícil y a veces no se pueden obtener muestras representativas.
Para desarrollar una estrategia de muestreo para desechos heterogéneos, primero es importante entender la escala, tipo y magnitud de la heterogeneidad.
IX.2 Tipos de heterogeneidad
La noción de heterogeneidad es relacionada con la escala de observación. Un ejemplo podría ser una pila de arena. Desde una distancia de pocos pies, una pila de arena parece ser uniforme y homogénea, sin embargo, a un rango bajo magnificación es heterogénea. Diferencias sustanciales son encontradas entre granos individuales en sus formas, tamaños, colores, densidades, dureza, composición del material, etc. Para algunos materiales, las diferencias entre granos individuales o partículas no son medibles o no son de relativa significancia para los objetivos del estudio.
A una escala de gran tamaño, como un sitio entero de desecho, rangos largos (escalas largas). Heterogeneidad a larga escala refleja tendencias locales y juega un importante papel en la decisión del uso de una evaluación para identificar patrones espaciales en el sitio, para usar diseños de muestreo estratificado para estimar un parámetro, o para definir los límites del problema de muestreo. Ítems, partículas o fases dentro de un desecho pueden ser distribuidas en varias vías para crear distintamente diferentes tipos de heterogeneidad. Estos tipos de heterogeneidad incluyen:
– Heterogeneidad al Azar: ocurre cuando ítems no similares son distribuidas de manera aleatoria por toda la población.
– Heterogeneidad no al Azar: ocurre cuando ítems no similares son distribuidas de manera no aleatoria, resultando en la generación de estrato. El término de estrato se refiere a subgrupos de una población separados en espacio, en tiempo, o por componentes remanentes de una población.
Las diferencias entre ítems o partículas que resultan de la heterogeneidad son debidas a las diferencia en su composición o propiedades. Una de esas propiedades-tamaño de la partícula-merece especial consideración porque significantes diferencias en el tamaño de la partícula son comunes y pueden complicar el muestreo debido al error fundamental. El error fundamental puede ser reducido solo a través de la reducción del tamaño de la partícula o la recolección de muestras suficientemente grandes.
Existen tres tipos de heterogeneidad que se describen en las normas ASTM: 1) Homogéneo, 2) Heterogéneo al azar, y 3) Poblaciones heterogéneas no al azar. Poblaciones dentro de barriles representan diferentes tipos de distribución espacial mientras que las poblaciones que vienen de una descarga representan tipos de distribución temporal.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura IX.1. Diferentes Tipos de Heterogeneidad Espacial y Temporal.
IX.3 Magnitud de heterogeneidad
Magnitud de heterogeneidad es el grado de diferencias en las características de interés en los fragmentos, partículas o volúmenes dentro de la población. Pueden clasificarse desde una población donde los ítems son muy similares, siendo prácticamente homogénea, a una población donde los ítems son muy diferentes. Medidas estadísticas de dispersión, la varianza y la desviación estándar, son indicadores útiles de los grados de heterogeneidad dentro de un desecho o un sitio.
Si el desecho exhibe heterogeneidad no al azar y una alta magnitud de heterogeneidad, entonces considerar como segregados (en el punto de generación) o el manejo del desecho como dos o más unidades separadas. Esta técnica puede requerir conocimiento previo de las porciones del desecho que caen dentro de cada categoría especificada (como escombro peligroso o no peligroso).
IX.4 Diseños de muestreo para desechos heterogéneos
La escogencia del adecuado diseño de muestreo para caracterizar desechos heterogéneos depende de los objetivos del estudio (identificación o clasificación del desecho, caracterización del sitio, etc.), los objetivos de los datos de calidad, el tipo y magnitud de heterogeneidad, y consideraciones prácticas como acceso a todas las porciones del desecho, seguridad y la disponibilidad de equipos para la obtención y preparación de las muestras.
Como se dijo anteriormente existen dos categorías de diseños de muestreo: Muestreo Probabilístico que se refiere a diseños de muestreo en el cual todas las partes del desecho o medio bajo un estudio tienen una probabilidad conocida de estar dentro de la muestra.
– Desecho heterogéneos al azar: Un diseño de muestreo probabilístico como una muestra al azar o un muestreo sistemático puede ser usado. Al menos una de las dos estrategias de recolección de muestras, sin embargo, también pueden ser usadas para mejorar la precisión del diseño de muestreo:
1. Tomar muestras individuales muy grandes, o
2. Tomar varios incrementos para formar cada muestra individual.
Desechos Heterogéneos no al azar: Una de las dos estrategias pueden ser utilizadas para mejorar el diseño de muestreo:
1. Si el objetivo es estimar un parámetro de la población general (como la distancia media) un muestreo al azar estratificado puede ser usado.
2. Si el objetivo es caracterizar cada estrato separadamente, una técnica apropiada para separar cada estrato en su punto de generación, y caracterizar y manejar cada estrato separadamente.
Si cualquier forma de muestreo estratificado es usada, entonces uno de los tres tipos de estratificación puede ser considerado:
– Estratificación por espacio.
– Estratificación por tiempo.
– Estratificación por componente.
< TABLA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura IX.2. Tres diferentes tipos de estrato.
Ejemplos de los tres tipos de estratificación
| Tipo de estratificación | Escenario ejemplo |
| Estratificación a través del espacio | Una acción de limpieza se requiere para remover 2 pies de sólidos en la parte superior del sitio. Antes de la excavación, el transportador del desecho desea conocer la concentración de los constituyentes del sólido que va a ser removido. Las 6 pulgadas superiores del sólido son conocidas para ser de mas alta contaminación que las 18 pulgadas de sólido remanente: el muestreo del sólido puede ser mas eficiente por la estratificación del sólido en dos subpoblaciones- la porción superior de 6 pulgadas y la porción de la parte inferior de 18 pulgadas |
| Estratificación a lo largo del tiempo | Una descarga de desechos desde varios tubos a lo largo del tiempo |
| Estratificación por componentes | Escombros cubiertos con pintura en la misma estructura con materiales como vidrio y madera pueden ser muestreados por componentes. |
IX.5 Técnicas de muestreo para desechos heterogéneos
Algunos métodos usados para muestreo de estructuras contaminadas o escombros son los siguientes:
– Cortar el escombro en piezas, después aplastarlo y molerlo, con esto puede tener un manejo en el laboratorio como una muestra sólida.
– Taladrar el molde seguido de la recolección de cortes para luego ser analizados. Esta técnica puede requerir 20 a 50 sitios de perforación en un área local para obtener un volumen suficiente.
– Moler una estructura entera en un tubo y después aplicar las técnicas convencionales de muestreo.
DISPOSITIVOS DE MUESTREO.
X.1 Bombas y sifones
Pueden ser usadas para obtener muestras de desechos líquidos y aguas subterráneas.
X.1.1 Muestreador automático.
Es un tipo de dispositivo de bombeo usado para la recolección de muestras periódicamente. Es recomendado para el muestreo de aguas superficiales y puntos de descarga. Este dispositivo puede ser usado en sistemas de recolección de aguas residuales y plantas de tratamiento e investigaciones de muestreo de corrientes. Se pueden encontrar, muestreadores automáticos para análisis de orgánicos volátiles.
Un muestreador automático usa bombas peristálticas como mecanismo de muestreo. Estas pueden ser programadas para obtener muestras en intervalos de tiempo o para obtener muestras continuas.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.1. Muestreador automático.
Ventajas:
– Puede proveer muestras simples o compuestas sobre el tiempo.
– Opera sin ser atendida, y puede ser programada para tomar muestras a volúmenes variables en tiempos variables.
Limitaciones:
– Requiere de energía eléctrica para su funcionamiento.
– Puede ser difícil su descontaminación.
– Puede no operar correctamente cuando existe alto porcentaje de sólidos en una corriente liquida.
X.1.2 Bomba bladder.
Recomendada para el muestreo de aguas superficiales, aguas subterráneas y puntos de descarga. También puede ser usada para el muestreo de líquidos en superficies comprimidas. Consiste de una membrana flexible encerrada por un contenedor y puede ser construida de una variedad de materiales como neopreno, caucho, acero inoxidable, nitrilo, etc. Existen dos tipos de bomba Bladder- tipo comprimido y tipo expandido. El de tipo apretado tiene el bladder conectada a la línea de descarga de la muestra. La cámara entre el bladder y la muestra es conectada a una línea de gas. El de tipo expandido tiene el bladder conectado a la línea de gas. En este tipo de bomba bladder, la cámara entre el bladder y la muestra es conectada a la línea de descarga de la muestra. Durante el muestreo, el agua entra al dispositivo mediante una válvula que está en el fondo del dispositivo. Aire comprimido o gas es entonces inyectado al dispositivo. Esto causa que el bladder se expanda o se comprima dependiendo del tipo de la bomba Bladder. La válvula se cierra y los contenidos del dispositivo son forzados para que entren a una línea de descarga. La parte superior de la válvula previene que el agua se devuelva. Por cambios de presión, el proceso es repetido para recolectar más muestras.
< TABLA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.2. Bomba Bladder.
Ventajas:
– El conveniente para el muestreo de líquidos conteniendo componentes volátiles.
– Puede recolectar muestras de hasta 60m de profundidad.
Limitaciones:
– La operación requiere grandes contenidos de aire comprimido o gas y un controlador.
– Requiere de una fuente de energía eléctrica.
– Puede ser pesado y difícil de operar.
– Su descontaminación puede ser difícil.
X.1.3 Bomba peristáltica. Es una bomba elevada de succión usada en la superficie para recolectar líquidos desde aguas subterráneas o superficies comprimidas. Puede ser usada para el muestreo de agua superficial, agua subterránea, puntos de descargas, líquidos comprimidos, y desechos líquidos de varias capas. Este dispositivo consiste de un rotor con rodillos almohadillados y este tiene una pieza de tubo flexible enroscada alrededor del rotor de la bomba y conectado a dos piezas de politetrafluroetileno u otro tubo disponible. El otro lado es conectado al contenedor de la muestra. Un tubo de silicona es comúnmente usado dentro de la cabeza de la bomba, sin embargo, para propósitos de muestreos orgánicos, silicona médica es recomendadble evitar la contaminación de la muestra.
Durante la operación el rotor aprieta el tubo flexible, causando un vacío para ser aplicado al tubo añadido. El material de muestra en atraído dentro del tubo añadido y descargado a través del final del tubo flexible. Para muestras liquidas en barriles, la bomba peristáltica es usada primeramente para mezclar la muestra. Ambas puntas del tubo son situadas en el contenedor y seguidamente se enciende la bomba. Una vez es terminada la mezcla, la muestra es recolectada como se describió anteriormente. Para la recolección de muestras para análisis de orgánicos volátiles el tubo unido al final de la bomba es llenado con la muestra y después es desconectado de la bomba.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.3. Bomba Peristáltica.
Ventajas:
– Puede recolectar muestras desde varias profundidades y diámetros pequeños.
– Fácil para usar.
– La bomba no requiere ser descontaminada. El tubo puede ser descontaminado o reemplazado.
Limitaciones:
– La formación del vacío puede provocar pérdidas de contaminantes volátiles.
– Profundidades para el muestreo no pueden exceder los 7.6 m.
– Requiere una fuente de energía eléctrica.
X.1.4 Bomba centrífuga sumergible. Es un tipo de bomba usada para purgar y monitorear el muestreo de pozos. Las partes que están en contacto con el agua pueden ser hechas de PTFE y acero inoxidable. La manguera de la bomba puede ser hecha de PTFE u otro material disponible. La cavidad del motor es llenada de aire o agua desionizada o destilada que puede ser reemplazada cuando sea necesario. Las tasas de flujo de la bomba van desde 100 ml. por minuto a 9 galones por minuto.
Durante la operación el agua es atraída por una leve succión creada por la rotación de los impulsadores. Los impulsadores trabajan contra los platos fijos y presuriza el agua, la cual es hacia la superficie a través de la manguera de descarga.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.4. Bomba Centrífuga Sumergib
Ventajas:
– Puede ser construida de materiales que son químicamente resistentes.
– Puede ser usada para bombeo de líquidos de hasta 75 m de profundidad.
– Las tasas de flujo son ajustables.
Limitaciones:
– Puede ser no compatible con líquidos que contengan alta concentración de sólidos.
– No es apropiada para la recolección de muestras para análisis de orgánicos volátiles.
X.1.5 Bombas de desplazamiento. Es un tipo de bomba para el muestreo de aguas superficiales, aguas subterráneas, puntos de descarga y otros tipos de líquidos. Esta bomba fuerza una columna de agua discreta hacia la superficie con una elevación mecánica. Durante la operación, el agua entra el dispositivo a través de una válvula que se encuentra en la parte inferior del dispositivo. Esta es comúnmente construida de PVC, acero inoxidable o ambas. Dos tipos comunes de bombas de desplazamiento incluyen la bomba de desplazamiento aire/gas y la bomba de desplazamiento pistón. El primer tipo de bomba de desplazamiento usa gas comprimido y opera con la aplicación positiva de presión a la línea de gas. Esto genera que la válvula de entrada se cierre y la válvula de la línea de descarga se abra, forzando el agua hacia arriba por la línea descarga hasta la superficie. El segundo tipo de bomba de descarga usa una barra accionadora desde la superficie o desde un activador eléctrico o de aire. El pistón es operado mecánicamente liberando la muestra a la superficie al mismo tiempo que la cámara es llenada.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.5. Bombas de Desplazamiento.
Ventajas:
– Puede ser construida de PTFE para reducir el riego de contaminación.
Limitaciones:
– Puede ser difícil su descontaminación.
– Requiere grandes volúmenes de aire o gas para su funcionamiento.
X.2 Dragados
Los sistemas de dragados incluyen equipos que se usan para recolectar material que se encuentra en el fondo de un contendedor (sedimentos) debajo de una capa de líquido en movimiento o un líquido estacionario. Una variedad de dragados son disponibles incluyendo el muestreador Ekman y el Dragador Ponar.
X.2.1 Dragador Ponar. Es recomendado para el muestreo de sedimentos. Este dispositivo tiene un par de mandíbulas que penetran el sustrato y se cierran para retener la muestra. El rango de volumen de la muestra es de 0.5 a 3.0 litros (ASTM D 6232).Es bajado lentamente con una velocidad controlada con tal de que el dispositivo se ubique adecuadamente y evitando que la superficie de muestreo sea removida. La poca tensión bloquea la abertura de las mandíbulas y permite que el dispositivo se cierre hasta que se llene en su totalidad. El dispositivo es subido lentamente para evitar disturbios en la muestra.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.6. Dragador Pon
Ventajas:
– Reutilizable.
– Puede obtener muestras de varios tipos de sedimentos estacionarios extendidos desde fango hasta material granular.
– Disponible en diferentes rangos de peso y tamaño.
Limitaciones:
– No adecuado para la recolección de muestras no perturbadas.
– Puede ser difícil su descontaminación.
– No puede recoger una muestra representativa repetidamente en varias ocasiones
a la misma profundidad y posición.
X.3 Muestreadores a alturas discretas
Son dispositivos que pueden recolectar muestras a alturas específicas.
X.3.1 Bomba Bacon. Es un tipo de muestreador a una altura discreta que provee una muestra desde una altura específica en un cuerpo estacionario de agua o desecho. Es recomendada para el muestreo de aguas superficiales y usualmente usada para recolectar muestras desde un lago o estanque. También se usa para recolectar muestras de desechos líquidos de grandes tanque o lagunas. El rango de volumen de la muestra es desde 0.1 a 0.5 litros (ASTM D 6232). Tiene un cuerpo cilíndrico, el cual es bajado dentro del material por una cuerda de soporte primaria y tiene un tapón interno que actúa como una válvula para admitir la muestra. Una segunda cuerda es atada en la parte superior del tapón para abrir y cerrar la válvula después del muestreo. La bomba permanece cerrada hasta que se acciona para recolectar la muestra. La recolección de la muestra es accionada por el levantamiento del tapón y permitiendo que el dispositivo se llene de la muestra. El dispositivo es entonces cerrado halando la cuerda del tapón. El dispositivo es retornado a la superficie por la cuerda primaria de soporte, y la muestra es transferida directamente al contenedor.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.7. Bomba Bacon.
Ventajas:
– Recolecta muestras a ciertas distancias, y no es abierta hasta alcanzar la altura deseada.
– Fácil uso.
Limitaciones:
– Puede ser difícil su descontaminación.
– Máxima capacidad de muestra es solo 500 ml.
– Materiales de construcción pueden no ser compatibles con parámetros de interés
X.3.2 Muestreador Kemmerer. Es un tipo de muestreador a una altura discreta que provee una muestra desde una altura específica en un cuerpo estacionario de agua o desecho. Es recomendada para el muestreo de aguas superficiales y usualmente usada para recolectar muestras desde un lago o estanque. También se usa para recolectar muestras de desechos líquidos de grandes tanques o lagunas. El rango de volumen de la muestra es de 1 a 2 litros.
El dispositivo comprende un brazo cilíndrico con tapones de cauchos en ambas esquinas. Las esquinas son dejadas abiertas mientras el dispositivo es bajado en una posición vertical, permitiendo un paso de agua o aire a través del cilindro. Cuando el dispositivo está a la altura deseada, un mensaje es enviado hacia para que los tapones sean cerrados. El dispositivo es entonces subido y la muestra es removida a través de una válvula hacia un contenedor.
Ventajas:
– Puede recolectar muestras a alturas discretas.
– Provee correcta delimitación y extracción de la muestra.
– Fácil uso.
Limitaciones:
– Puede ser difícil su descontaminación.
– El muestreador se expone al medio en otras profundidades mientras que es bajado a un punto de muestreo en la profundidad deseada.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.8. Muestreador Kemmerer.
X.3.3 Muestreador tipo jeringa. En un dispositivo para muestrear líquidos a alturas discretas. Con la opción de la base extrema, puede ser utilizado como dispositivo de base para muestrear líquidos altamente viscosos, lodos, y sustancias grumosas. Es usado para la recoleccione de muestras desde barriles, tanques, y superficies comprimidas, también puede obtener muestras cuando solo una cantidad pequeña permanece en el fondo del tanque o barril. El rango de volumen de la muestra es de 0.2 a 0.5 litros.
Este dispositivo es construido de un pistón que comprime una manilla en forma de T, una tuerca de seguridad, una barra de control, un montaje con cuerpo de pistón, un montaje de tubo de muestreo y dos extremidades. Cuando se usa como jeringa, el dispositivo es bajado al punto de muestreo y la manivela en forma de T es gradualmente levantada para recolectar la muestra. Una vez se ha obtenido la muestra deseada, la tuerca de fijación se aprieta para asegurar la barra pistón y la válvula inferior es cerrada apretando muestreador contra el lado o el fondo del envase. Cuando es usado como un dispositivo en una base, el dispositivo es lentamente introducido dentro del material. Cuando se tiene la muestra deseada, la tuerca de fijación se aprieta para asegurar la barra pistón y el dispositivo se retira del medio. El material de muestra es sacado dentro del contenedor abriendo la válvula inferior, aflojando la tuerca de fijación, y presionando el pistón hacia abajo.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.9. Muestreador Tipo Jeringa.
Ventajas:
– Es fácil de usar y descontaminar.
– Puede muestrear fondos de contenedores.
Limitaciones:
– Se puede utilizar a profundidades de cerca de 1.8 metros solamente.
X.3.4 Muestreador agua/lodo tapado. Es un dispositivo que proporciona muestras a alturas específicas. Es usado para recolectar lodos o desechos sólidos provenientes de tanques, camiones con tanque, y estanques. Puede muestrear líquidos, desechos líquidos de varias capas, y desechos con fases con mezclas sólidas/líquidas. El volumen de muestra típico es 1.0 litro.
Comprende una jarra de vidrio desprendible, algunas veces con un cortador para materiales con contenidos de más de 40% de sólidos que es montado en un dispositivo de acero inoxidable.
El dispositivo es introducido dentro del material que va a ser muestreado y abierto a la altura deseada. La manivela superior es rotada verticalmente a la jarra y abre y cierra la tapa. El dispositivo es retirado del material, la jarra es removida del dispositivo levantándolo desde el contenedor, y la jarra sirve como contenedor de la muestra.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.10. Lidded Sludge/Water Sampler
Ventajas:
– La jarra del dispositivo también sirve como contenedor.
– Las botellas y las tapas son únicas en cada muestra, por lo tanto, la descontaminación de un envase no se requiere.
Limitaciones:
– Pesado y limitado a un tamaño de la botella.
– El lodo grueso es difícil de muestrear.
X.3.5 Muestreador a niveles discretos. Es un muestreador cilíndrico desmontable ajustada con una válvula operada manualmente. Es recomendado para el muestreo de aguas superficiales, aguas subterráneas, puntos de descarga, líquidos, líquidos con múltiples capas, y es usado para el muestreo de tanque, contenedores, pozos, y superficies comprimidas. El rango de volumen de muestra típico es de 0.2 a 0.5 litros. Este dispositivo está compuesto de un tubo ajustado con válvulas operadas manualmente, las cuales son operadas por un montaje de control atado a la parte más alta del dispositivo. Este montaje consiste de un tubo rígido y una vara o tubo flexible y una cuerda interna.
Para recoger una muestra, el dispositivo se introduce dentro del material de la muestra a la profundidad de muestreo deseada. La válvula o las válvulas son abiertas manualmente para recoger la muestra y se cierra antes de recuperar el dispositivo. El modelo estándar es vaciado presionando la válvula de descarga contra el lado del envase de muestra. El muestreador de válvula dual es vaciado abriendo las válvulas manualmente. Alternativamente, la muestra recogida se puede llevar al laboratorio en el dispositivo substituyendo las válvulas por tapas.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.11. Muestreador a Niveles Discretos.
Ventajas:
– Relativamente fácil de descontaminar.
– Puede ser usado para muestras líquidas en la mayoría de las situaciones ambientales.
– Puede ser operado en ambientes peligrosos.
– El cuerpo del dispositivo puede ser usado como dispositivo de almacenamiento y transporte.
Limitaciones:
– Limitado para muestrear capacidades de 240-475 ml.
– Puede ser incompatible con los líquidos con alto porcentaje de sólidos
X.4 Dispositivos a presión
X.4.1 Muestreador penetrante. Es un dispositivo empujador, por lo tanto, provee un núcleo de muestra. Es recomendado para el muestreo de sólidos. El rango de volumen de la muestra es de 0.2 a 2.0 litros.
Consiste de uno o varios tubos de acero roscados, una tapa superior, y una extremidad de acero desmontable. Los tubos de acero tiene un diámetro de 1 pulgada o de menos longitud.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.12. Muestreador Penetrante.
Ventajas:
– Fácil de descontaminar y es reutilizable.
– Puede proveer muestras para análisis en el sitio de muestreo.
– Versátil y puede muestrear 15 a 20 localizaciones al día para cualquier combinación de matrices
Limitaciones:
– Puede ser pesado y voluminoso dependiendo del tamaño usado.
– Limitado por la composición de materiales subterráneos y accesibilidad a materiales más profundos.
– Puede ser inadecuado para los materiales que requieren fuerza mecánica para su penetración.
X.4.2 Muestreador (Split Barrel Sampler). Es un dispositivo empujador frecuentemente usado con un equipo de taladro para recoger muestras subterráneas. Los materiales que se muestrearán debe ser bastante húmedos permanecer en el muestreador. El rango de volumen de muestra es de 0.5 a 30 litros.
Consiste de un tubo largo de acero dividido longitudinalmente y equipado con un impulsador hecho de acero y un conductor. El impulsador es desprendido y puede ser reemplazado deformado. Su longitud es típicamente de18 a 30 pulgadas y su diámetro interior es de 1.5 a 2.54 pulgadas. Puede ser usado para tomar muestras sólidas no alteradas a alturas considerables.
Este dispositivo es situado sobre la superficie del material que va a ser muestreado, empujándolo hacia abajo mientras que se tuerce levemente. Un taladro puede ser adherido al equipo, ya que la presión a mano algunas veces es difícil. Cuando la altura es alcanzada, este dispositivo es torcido para partir el centro de la muestra, entonces el dispositivo es retirado del material. La muestra puede ser removida desde el barril o el medio de transporte para el análisis. Con una cuerda, la muestra puede ser removida con una mínima cantidad de disturbios.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.13. Split Barrel Sampler.
Ventajas
– Reutilizable, fácil de descontaminar y de usar.
– Proporciona una muestra relativamente imperturbada, por lo tanto, puede reducir al mínimo la pérdida de compuestos orgánicos volátiles.
Limitaciones:
– Requiere un taladro o un aparato de empuje para las muestras profundas.
X.4.3 Tubo hueco concéntrico. Es un dispositivo de empuje que en donde el usuario empuja directamente el material que se muestreará. Puede ser usado para muestras sólidas arenosas o granulares y desechos en pilas o bolsas, tanque o contenedores similares. Los tubos concéntricos son generalmente de 61 a 100cm de longitud y 1.27 a 2.54 cm de diámetro. El rango de volumen de la muestra es de 0.5 a 1.0 litros.
Consiste en dos tubos con ranuras. El tubo más exterior tiene un extremo puntiagudo cónico en uno de sus extremos el cual permite que el tubo penetre el material que está siendo muestreado. El tubo es abierto y cerrado por la rotación del tubo interior, y es insertado dentro del material cuando esta cerrado. Una vez insertado, el tubo interior es rotado abiertamente y el dispositivo es meneado para permitir que el material entre por las aberturas. Después el tubo es cerrado y retirado.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.14. Tubo Hueco Concéntrico.
Ventajas:
– Es un buen muestreador de presión directa para materiales secos no consolidados.
– Fácil de usar.
Limitaciones:
– Puede ser difícil su descontaminación.
– No recomendado para el muestreo de materiales húmedos o pegajosos.
– No recolecta muestras que contengan todos los tamaños de partícula si el diámetro de la partícula sólida más grande es una mitad mayor que del ancho de la ranura.
X.4.4 Trier. Es un dispositivo de empuje que asemeja una cuchara alargada y es usada para muestrear sólidos húmedos o pegajosos con un diámetro de partícula menor que una mitad de una porción de diámetro del tubo. Puede ser utilizado para muestrear suelos y materiales cohesivos de grano fino similares. El volumen de rango típico de la muestra es de 0.1 a 0.5 litros.
Consiste de una manivela conectada a un tubo cortado longitudinalmente, con una extremidad afilada que permite cortar el material.
Típicamente son de 61 a 100cm de longitud y 1.27 a 2.54 cm. de diámetro, usados como un dispositivo de corte vertical, donde una muestra cilíndrica o relativamente completa puede ser extraída.
El dispositivo es empujado dentro del material que va a ser muestreado y girado una o dos veces para cortar el corazón del material. La rotación se para con la abertura abierta señalada hacia arriba. El corazón es removido del hueco, evitando que el material de la sobrecarga se convierta en una parte de la muestra.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.15. Traer.
Ventajas.
– Es un buen muestreador de presión directa para materiales húmedos o pegajosos.
– Liviano, fácil de usar y de descontaminar.
Limitaciones:
– Limitado para muestrear tamaños de partícula dentro del diámetro del tubo insertado y no recogerá partículas de mayor tamaño que la anchura de la ranura.
– No recomendado para el muestreo de sólidos secos no consolidados.
– Solamente el muestreo superficial, y la profundidad de la muestra es limitado por la longitud del tubo.
X.4.5 Tubo de paredes delgadas. Es un dispositivo para el muestreo de sólidos cohesivos, no consolidados, particularmente suelos. No se recomienda para grava o suelo rocoso. El rango del volumen de muestra es de 0.5 a 5.0 litros. Tiene comúnmente 30 pulgadas de largo y con diámetros exteriores de 2, 3, y 5 – pulgadas.
El tubo se une con tornillos de presión a una longitud de una barra sólida o tubular, y el extremo superior de la barra, o la cabeza del muestreador, es enroscada para aceptar una manija o una barra de extensión. El tubo puede ser utilizado conjuntamente con los taladros del mismo tamaño.
El extremo del muestreador se empuja directamente al medio usando una fuerza hacia abajo sobre la manivela. Puede ser empujado hacia abajo con un gato o con un pistón hidráulico. Una vez la profundidad deseada se ha alcanzado, el tubo se tuerce para romper la continuidad del extremo y se retira del medio. El material de la muestra se saca en el envase de muestra forzando una barra a través del tubo.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.16. Tubo de Paredes Delgadas.
Ventajas:
– Disponible fácilmente, económico, y fácil de utilizar.
– Reutilizable y puede ser descontaminado.
– Obtiene una muestra relativamente imperturbada.
Limitaciones:
– Algunos tubos de paredes delgadas son grandes y pesados.
– El material que se muestreará debe ser de una consistencia física (material cohesivo) para ser retirado dentro del tubo. No puede ser utilizado para muestrear grava o suelos rocosos.
– Una pérdida volátil es posible cuando la muestra se quita del tubo.
– Los materiales con partículas de tamaño más grande de una mitad del diámetro interno del tubo no se deben muestrear con un tubo de paredes delgadas.
X.4.6. Muestreador tipo base (con válvula). Es un tipo de dispositivo de base empujador recomendado para suelo mojado, y puede también ser usado para muestrear desechos sólidos no consolidados, desechos con fases sólida/líquida, y pulverizados. Este dispositivo puede ser usado en tanques y pequeños contenedores como también en tanques, lagunas, y superficies comprimidas. El rango del volumen de la muestra es de 0.2 a 1.5 litros.
El muestreador tipo base con válvula es un muestreador cilíndrico de acero inoxidable con una extremidad como base, una tapa superior, una extensión con una manija cruzada, y una válvula de no retorno en el extremo inferior. La válvula es un dispositivo de retención para almacenar la muestra en el sitio hasta que el dispositivo sea retirado. Las muestras son normalmente en un transportador opcional. Este dispositivo es operado por la unión de una manivela o una extensión con una manija a la tapa del dispositivo. El dispositivo es bajado a la superficie, introduciéndolo dentro del material muestreado y removido.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.17. Muestreador Tipo Base.
Ventajas:
– Reutilizable y es descontaminado fácilmente.
– Proporciona una muestra relativamente imperturbada.
– Puede ser operada manualmente y no requiere fuerza física significativa.
Limitaciones:
– No puede ser utilizado en grava, grandes partículas de sedimentos, o lodos.
– Cuando el muestreo se realiza para compuestos orgánicos volátiles, se debe utilizar con un trazador y una cápsula para reducir al mínimo la pérdida de volátiles.
X.4.7 Muestreador de base miniatura. Puede ser usado para recolectar sólidos y muestras de desechos para análisis de compuestos orgánicos volátiles. Este tipo incluye dispositivos como el Muestreador Sólido atrapado/purgado™, el muestreador EnCore™, o una jeringa plástica. Este dispositivo también puede ser usado como envase hermético del almacenaje y contenedor de la muestra para su envío.
Este dispositivo recolecta una pequeña submuestra y es particularmente útil para el muestreo de análisis de componentes orgánicos volátiles.
Es recomendado para el muestreo de sólidos desde el suelo o del lado de una fosa, y puede ser utilizado para el muestreo de sedimentos y desechos sólidos no consolidados. No puede ser utilizado para el muestreo de material cementado, material consolidado, o material que tiene fragmentos bastante gruesos. El muestreador EnCore™ puede ser utilizado para recolectar submuestras de corazones de sólidos y tiene un rango de volumen de muestra de 0.01 a 0.05 litros. El dispositivo está disponible en dos tamaños para la colección de muestras de 5 y 25 gramos. La escogencia del dispositivo se basa en el tamaño de muestra requerido por el procedimiento analítico.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.18. Muestreador Base Miniatura.
Ventajas:
– Mantiene la estructura de la muestra en un dispositivo que también se puede utilizar para almacenar y para transportar la muestra directamente al laboratorio.
– Recomendado para recoger muestras para el análisis de compuestos volátiles. Recoge una muestra relativamente no perturbada que es contenida antes del análisis para reducir al mínimo la pérdida de compuestos volátiles.
– Es generalmente compatible con los productos químicos y las características físicas de los medios muestreados.
– Ningunas limitaciones físicas significativas para su uso.
Limitaciones:
– No puede ser utilizado para muestrear grava o suelos rocosos.
– Las instrucciones se deben seguirse cuidadosamente para su uso apropiado para evitar que el aire choque con la muestra y asegurar que la muestra no comprometa los tapones.
X.4.8 Muestreador de jeringa modificada. Es un dispositivo para muestreo empujador construido de una jeringa modificada. Puede ser utilizado para proveer submuestras de suelos, sedimentos y desechos sólidos no consolidados, también sirve para submuestras de tamaños grandes de suelos. NO es recomendado para el muestreo de sólidos cementados, material consolidado o material con fragmentos de arena gruesa. A diferencia del muestreador EnCore ™, este dispositivo no puede ser usado para almacenar no para transportar la muestra al laboratorio.
También sirve para muestrear componentes volátiles. El rango de volumen de este dispositivo es de 0.01 a 0.05 litros.
Se construye por el corte del parte extremo inferior del accesorio de la jeringuilla unida de la aguja. El émbolo es removido del tapón de caucho, y el émbolo se empuja hacia adentro hasta alcanzar el mismo nivel con el extremo del corte. El muestreador es introducido para recolectar la muestra, la cual puede ser dispuesta en un vaso VAO para su almacenamiento y posterior transporte al laboratorio. La muestra es introducida dentro del contenedor presionando el tapón de la jeringa.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.19. Muestreador Jeringa Modificada.
Ventajas:
– Obtienes perfiles de muestra no perturbadas.
– Puede ser usado para la recolección de muestras para el análisis de componentes orgánicos volátiles.
– No tiene limitaciones físicas para su uso.
– Económico.
Limitaciones:
– No puede ser utilizado para muestrear la grava o suelos rocosos.
– El material de la construcción puede ser incompatible con medios altamente contaminados.
– Es necesario asegurarse que el dispositivo esté limpio antes de usarlo.
X.5 Dispositivos de bases rotadoras.
Incluyen equipos que obtienen columnas verticales de una muestra de sólido por medio de una acción rotativa. Algunos de estos dispositivos (como taladros) también se pueden utilizar para abrir un agujero para la colección de la muestra a cierta profundidad usando otra pieza de equipo.
X.5.1 Taladro Bucket. Es un dispositivo de base rotativa operado manualmente generalmente usado para el muestreo de sólidos, sedimentos, o desecho sólidos no consolidados. Puede ser usado para obtener muestras en tanques, contenedores de almacenamiento, y pilas de desecho. El rango de volumen de muestra es de 0.2 a 1.0 litros.
La cabeza cortada del taladro es introducida y torcida con la mano con una fuerza descendiente dentro del suelo y removida cuando el dispositivo está lleno. El taladro vacío es retornado al hueco y el procedimiento es repetido. El procedimiento es repetido hasta que se logra la altura deseada. Este dispositivo puede ser usado para aumentar el hueco si es una muestra compuesta de varios intervalos de tiempo; sin embargo, muestras discretas simples pueden ser recolectadas en taladros separados.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.20. Taladro Bucket.
Ventajas.
– Reutilizable y fácil de descontaminar.
– Efectivo para muestras subterráneas pocas profundas.
– Permite el uso de varias cabezas de taladro para muestrear sólidos a diferentes condiciones.
Limitaciones:
– La altura de muestreo es limitada sobre 20 pies (6m) debajo de la superficie.
– No útil para muestras no alteradas.
– No ideal para el muestreo de componentes orgánicos volátiles.
X.5.2 Dispositivo de base giratoria. Es un dispositivo que recolecta columnas verticales de muestras de sólidos a través de una acción giratoria y puede ser usada para el muestreo de desechos sólidos consolidados, suelos, y sedimentos. El rango de volumen de la muestra es de 0.5 a 1.0 litros.
Consiste de un cilindro de acero inclinado unido a un taladro eléctrico. El dispositivo puede ser operado por un taladro manual o con taladro montado en un stand. La longitud del barril es usualmente de 1 a 1.5 pies y los rangos del diámetro van de 2 a 6 pulgadas. Puede ser usado para muestras a una altura específica o muestras superficiales.
Es situado verticalmente a la superficie del medio que va a ser muestreado, y encendido antes de estar en contacto con la superficie. Una presión continua y uniforme es suministrada al dispositivo hasta que la altura deseada es alcanzada. Una vez lograda la altura, el dispositivo es retirado y la muestra es colocada dentro de un contenedor para el análisis.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.21. Dispositivo de Base Giratoria.
Ventajas:
– Fácil de descontaminar.
– Reutilizable.
– Puede obtener corazones de muestras sólidas.
Limitaciones:
– Requiere de batería u otra fuente de energía.
– Requiere un suplemento de agua, usada para refrescar el tubo.
No es fácil para operar.
X.6 Dispositivos para perfiles líquidos
Son dispositivos que incluyen equipos que pueden recolectar una columna vertical de un líquido, lodo o mezclas de líquidos.
X.6.1 Coliwasa (muestreador de desechos líquidos compuestos). Es usado para obtener una columna vertical del material muestreado. Es recomendado para el muestreo de líquidos, desechos con líquidos de múltiples capas, y desechos con mezclas sólido/líquido y es comúnmente usado para el muestreo de líquidos en contenedores, como tanques y barriles. También sirve para muestrear líquidos estancados.
El rango de volumen de muestra es de 0.5 a 3 litros. Puede ser construido de PVC, vidrio, metal, PTFE o cualquier otro material compatible con la muestra que va a ser tomada. En general, un Coliwasa consta de un tubo con un final tapado y una varilla en su interior que tiene en algunos tipos tapones en las extremidades. Una configuración consta de una válvula pistón atada a una varilla interna para bloquear el mecanismo en el otro extremo. Coliwasa están disponibles en varios diámetros (0.5 a 2 pulgadas) y longitudes (4 a 20 pies).
El Coliwasa es bajado lentamente a un ángulo exacto con la superficie del material. El dispositivo es abierto en ambas extremidades mientras es bajado para permitir que el material entre. Cuando se llega a la altura deseada, el muestreador es cerrado y ambos tubos son removidos del material. El material tomado es entonces transferido a un contenedor.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.22. COLIWASA.
Ventajas:
– Provee correcta relimitación y extracción del desecho.
– Fácil de usar.
– Económico.
– Reutilizable.
Limitaciones:
– Si es de vidrio puede quebrarse.
– La descontaminación puede ser difícil.
– El tapón puede no permitir la recolección del material en el fondo de un barril.
– Fluidos con alta viscosidad son difíciles para muestrear.
X.6.2 Tubo hueco. Es un dispositivo para la toma de muestras de perfiles líquidos, abierto en las extremidades que proporciona una columna vertical del material muestreado.
Es recomendado para el muestreo de líquidos, desechos líquidos de múltiples capas, y desechos líquidos con mezclas sólido/líquido y puede ser usado para tomar muestras de líquidos en barriles o contenedores similares. Pueden ser hechos de vidrio, acero inoxidable, o cualquier otro material disponible. Son típicamente de 48 pulgadas de longitud y de 6mm a 16mm de diámetro interno. Para muestras líquidas con baja tensión superficial, un bailer estrecho es más adecuado. Para muestras de lodos es mejor el uso de tubos pares. El tubo hueco es bajado verticalmente dentro del material, insertado lentamente para permitir que el nivel dentro y afuera del tubo sea el mismo. Esto evita incorrectas proporciones de muestras. La extremidad superior es sellada con un tapón para mantener la muestra en el tubo mientras es removida al contenedor.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.23. Tubo Hueco.
Ventajas:
– De fácil uso y económico.
Disponible en modelos desechables y no desechables.
Limitaciones:
– La altura de muestreo es limitada a la longitud del dispositivo.
– Puede no recolectar muestras en el fondo de un barril. La profundidad del material no muestreado depende de la densidad, tensión superficial, y viscosidad del material.
– Materiales con viscosidad alta son difíciles de muestrear.
– Puede ser difícil retener la muestra en el tubo cuando los líquidos muestreados tienen una alta gravedad específica.
X.6.3 Muestreador tubo valvulado. Es un dispositivo para la toma de muestras de perfiles líquidos, que proporciona una columna vertical del material muestreado. Es recomendado para el muestreo de líquidos, desechos líquidos de múltiples capas, y desechos líquidos con mezclas sólido/líquido y puede ser usado para tomar muestras de líquidos en barriles o contenedores similares. El rango de volumen de muestra es de 0.3 a 1.6 litros.
Puede ser construido de PTFE para reuso o polipropileno para un solo uso y consta de un tubo equipado con un tapón en la parte superior y una válvula en la parte inferior. Una argolla indicadora resbaladiza permite identificar niveles específicos o interfaces en los fluidos. El dispositivo es abierto en ambas extremidades durante la recolección de la muestra y bajado verticalmente dentro del material. El dispositivo es introducido lentamente para igualar los niveles dentro y fuera del tubo. Una vez se ha recolectado la cantidad de muestra deseada, el tapón y la parte inferior son cerrados. La parte superior es cerrada manualmente y la válvula en la parte inferior es cerrada aplicando una presión.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.24. Muestreador Tubo Valvulado.
Ventajas:
– De fácil uso, económico, e irrompible.
– Obtiene muestras a alturas alrededor de 8 pies (2.4 m).
Limitaciones:
– Algunas veces difícil de descontaminar
– Líquidos con alta viscosidad pueden ser difíciles de muestrear
X.6.4 Muestreador tipo émbolo. Es un dispositivo de muestreo de perfiles líquidos usado para recolectar columnas verticales de líquidos y es recomendado para el muestreo de líquidos con una o varias capas o mezclas de sólidos y líquidos. Puede ser usado para recolectar muestras en barriles, superficies comprimidas y tanques. El volumen de las muestras es de por lo menos 0.2 litros, y depende en el tamaño del contenedor de la muestra.
Este dispositivo consta de un tubo de muestra, una varilla o cuerda de muestra, una sección superior y un émbolo y está hecho de HDPE, PTFE, o vidrio. Una jarra para la muestra es conectada en la sección superior. El tubo es introducido dentro del líquido a la altura deseada. El émbolo es acoplado dentro del tubo para asegurar la muestra dentro del tubo y la cuerda o la varilla es levantada para transferir la muestra directamente dentro de la jarra.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.25. Muestreador Tipo Embolo.
Ventajas:
– Fácil de usar.
– Provee un sistema de recolección sellado.
– Disponible en varias longitudes.
Limitaciones:
– Cuando se usa tubo de vidrio es indispensable tener cuidado.
– Su descontaminación puede ser difícil, particularmente cuando se usa un tubo de vidrio.
X.6.5 Perfilador de sólidos fijos (lodo fijo). Es usado para medir o muestrear sólidos fijos (suspendidos) encontrados en plantas de tratamiento de aguas residuales, estanque de desechos y desechos con contenidos comprimidos. El rango de volumen de muestra es de 1.3 a 4.0 litros.
Es hecho de PVC y tiene marcas cada pie en sus 5 pies secciones de su cuerpo. Tiene una válvula en su sección más baja y una cuerda en la parte superior y es unida usando las uniones de las secciones para lograr la longitud deseada para el muestreo. El dispositivo es introducido dentro del medio hasta que este se llene. El nivel de los sólidos fijos puede ser medido por las marcas del tubo.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.26. Perfilador de Sólidos Fijos.
Ventajas:
– Permite medir columnas de líquidos y sólidos fijos a cualquier altura.
– Fácil de armar y usar.
– Reutilizable.
Limitaciones:
– Pueden ser difícil el muestreo de material con alta viscosidad.
X.7 Dispositivos para muestreos superficiales
X.7.1 Bailer. Son diseñados para obtener muestras de aguas subterráneas; sin embargo, también se pueden usar para obtener muestras de líquidos y desechos líquidos de varias capas en tanques y superficies comprimidas. No son útiles para el muestreo de lodos. El volumen de muestra es de 0.5 a 2 litros.
Un bailer es un tubo hueco con una válvula chequeadota en la base o válvulas en ambas extremidades. Un bailer puede ser unido en el medio con tubos para lograr su extensión y aumenta el volumen de la muestra. Están construidos de acero inoxidable, PVC, PTFE, o cualquier otro material disponible y esta disponible en varios tamaños. El bailer es atado a una cuerda y es bajado a la muestra. Mientas el tubo es bajado, la válvula en el fondo del tubo permite que el agua entre dentro del tubo. Después el tubo es retirado lentamente hacia la superficie. El peso del agua cierra la válvula.
Ventajas:
– De fácil uso, económico, y no requiere de una fuente de energía.
– Se puede construir de cualquier material.
– Relativamente fácil de descontaminar entre cada toma de muestra.
Limitaciones:
– No diseñado para obtener muestras desde alturas específicas debajo de líquidos superficiales.
– Si se baja rápidamente, puede perturbar la columna de agua.
– Contenidos altos de sólidos suspendidos o temperaturas altas pueden perturbar la operación de la válvula.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.27. Bailer.
X.7.2 Cucharón. Es un tipo de dispositivo de muestreo superficial usado para tomar muestras superficiales de barriles, superficies comprimidas, tanques, tubos, y puntos de descarga. Los puntos de muestreo son de poca profundidad (10 pulgadas) y tomadas sobre o debajo de la superficie. El rango de muestra es de 0.5 a 1.0 litros.
Este dispositivo consta de un beaker plástico, metálico o de vidrio sujetado al extremo de un palo largo de aluminio o fibra de vidrio, el cual sirve como un manubrio. Es usado por el sumergimiento del beaker dentro del material. El beaker es llenado y girado, después se retira del medio.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.28. Cucharón.
Ventajas:
– Económico.
– Fácil de manejar.
Limitaciones:
– No apropiado para el muestreo de capas subterráneas.
– Solamente se puede usar para recolectar muestras superficiales.
X.7.3 Muestreador de líquidos simples. Recolecta muestras a pocas profundidades debajo de superficies líquidas. Puede ser usado para recolectar líquidos o lodos en superficies comprimidas, tanques y barriles. El volumen de muestra es de 0.5 a 1.0 litros. El dispositivo es hecho de polipropileno o PTFE con un manubrio de acero inoxidable. La jarra muestreadora es usualmente hecha de vidrio. Esta se introduce dentro de la muestra y allí se abre la válvula para que la jarra se llene. La válvula se cierra antes de retirar el dispositivo.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.29. Muestreador de Líquidos Simples.
Ventajas:
– De fácil uso.
– La jarra puede ser tapada y usada como medio de transporte, además minimiza la pérdida de compuestos orgánicos volátiles.
Limitaciones:
– Cuando la jarra es de vidrio se requiere de mucho cuidado para evitar el rompimiento.
– Materiales de construcción necesitan ser compatible con el medio muestreado.
X.7.4 Muestrador oscilatorio. Recolecta muestras superficiales a distancias alrededor de los 12 pies. Puede ser usado para el muestreo de diferentes tipos de unidades, incluyendo barriles, superficies comprimidas, tanques, tubos/puntos de descarga, puertos de muestreo, y sitios de almacenamiento. Este tiene un rango de volumen de 0.5 a 1.0 litros.
Es normalmente usado con jarras muestreadoras de polietileno de alta densidad y tiene un manubrio de aluminio extensible con un pivote en la unión del manubrio con la jarra. El pivote permite recolectar muestras a diferentes ángulos.
Ventajas:
– De fácil uso.
– Recolecta muestras a varios ángulos.
Limitaciones.
– No puede recolectar muestras a discretas alturas.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.30. Muestreador Oscilatorio.
X.7.5 Cucharas, cucharones, paletas y palas. Son dispositivos de muestreo superficial usados para el muestreo de lodos, sólidos, polvo y desechos sólidos. El rango típico de muestra es de 0.1 a 0.6 litros para cucharas o cucharones y 1.0 a 5.0 litros para palas. Da mejores resultados cuando el material es uniforme.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura X.31. Cucharas, cucharones, paletas
Ventajas:
– Es uno de los dispositivos preferidos para el muestreo unidimensional de sólidos.
– Fácil de descontaminar y reutilizables.
– Económicas.
Limitaciones:
– Limitadas en superficies pocas profundas.
– Pueden causar la perdida de compuestos orgánicos volátiles.
PROCESO DE LIXIVIACIÓN PARA MATERIALES DE DESECHO MONOLÍTICOS “ENSAYO DE TANQUE” (ENSAYO DE DIFUSIÓN).
Este método es una traducción parcial de la norma EA NEN 7375:2004 de la Agencia Ambiental del Reino Unido, que esta basado en una traducción de la Norma Holandesa NEN 7375 (2004) del Instituto de Estandarización “Leaching characteristics-Determination of the leaching of inorganic components from moulded or monolithic materials with the difusion test – solid earthy and stony materials”.
X. ALCANCE Y APLICABILIDAD
El método original tiene como objetivo determinar la lixiviación de compuestos inorgánicos de materiales de desecho moldeados o monolíticos, usando el ensayo de difusión referido como ensayo de tanque para determinar la aceptación del desecho en rellenos. Sin embargo en este caso se va a utilizar para evaluar la característica de corrosividad por pH.
El propósito del ensayo de difusión es determinar la lixiviación de componentes inorgánicos de materiales moldeados y monolíticos bajo condiciones aerobias.
Este ensayo no es adecuado para materiales que son solubles durante la escala de tiempo del ensayo.
Este método presenta la forma de obtener el lixiviado de materiales de desecho moldeados o monolíticos, la determinación del pH se deberá realizar siguiendo el procedimiento específico para esta determinación.
Debido a que el tiempo total del ensayo es de 64 días, se sugiere una evaluación corta a 4 días (esta misma recomendación es admitida por Comunidad Económica europea para la evaluación de los criterios de aceptación para la disposición de residuos en los diferentes tipos de rellenos).
XI. PRINCIPIO DEL METODO
El método simula la lixiviación de componentes inorgánicos bajo condiciones aerobias en un período de tiempo de 64 días. El ensayo determina la naturaleza y propiedades del material bajo investigación localizando una muestra completa en un fluido de lixiviación (agua pH neutro, desmineralizada), reemplazando el eluato en tiempo especificados. Se determinan las concentraciones de los componentes del lixiviado en las fracciones del eluato sucesivas. El pH al cual el lixiviado se genera es determinado por el propio material.
XII. INTERFERENCIAS
No se especifican.
XIII. EQUIPOS Y MATERIALES
Todos los equipos deben ser verificados antes de usarse para asegurar su apropiada operación y la ausencia de interferencias que puedan afectar los resultados del ensayo. Estos no deben emitir, ni absorber cualquier componente que vaya a ser determinado en el eluato.
Piezas de ensayo: El ensayo de difusión requiere al menos una pieza a ensayar con la estructura, homogeneidad y composición que sea representativa del material o producto que se va a evaluar. La menor dimensión de la pieza de ensayo (P) debe ser mayor de 4 cm y se debe conocer el volumen [Vp] en litros.
Si el material que va a ser probado es producido en un formato de producto del cual la menor dimensión es menor de 4 cm, este producto solo puede ser usado como una pieza de ensayo si uno de los lados tiene un área superficial geométrica [A] mayor de 75 cm2.
Nota: Para incrementar la representatividad del material de ensayo, se acepta agrupar un número de piezas de un bache para el ensayo de difusión. El volumen [Vp] y el área superficial geométrica [A] se toma como el volumen total y el área superficial geométrica total de las piezas colectivas.
Tanque sellable: Tanque de plástico sin agentes suavizantes de volumen entre dos y cinco veces el volumen [Vp] y de dimensiones tales que la pieza del ensayo este rodeado al menos por 2 cm de agua por cada uno de los lados.
Equipo de filtración: Equipo de filtración adecuado para la filtración a alta o baja presión, el cual es enjuagado consecutivamente con ácido nítrico de concentración 1 ± 0,1 mol/l.
Filtros de membrana: Filtros con tamaño de poro de 0,45 µm.
pHmetro: Equipo calibrado, con una exactitud mejor de ± 0,05 pH unidades.
Beaker o balanza: Recipiente con una capacidad de medición de al menos seis veces el volumen Vp de la pieza de ensayo y una exactitud en la medida mayor a ± 1%, o una balanza con una capacidad de al menos tres veces el peso de la pieza de ensayo y una exactitud mayor de ± 0,1%.
XIV. REACTIVOS
Agua desmineralizada con una conductividad máxima de 1 µS/cm.
Acido nítrico de grado analítico en una concentración de 1 ± 0,1 mol/l.
XV. PROCEDIMIENTO
Los pasos para el ensayo son:
Determinación área geométrica de la pieza a ensayar.
Comportamiento del ensayo de difusión.
Análisis del eluato.
VI.i. Determinación del área geométrica [A] de la pieza a evaluar
El área geométrica de la pieza es determinada midiendo los parámetros característicos del área superficial geométrica.
2. Determinación del área geométrica de piezas regulares con una dimensión mayor a 4 cm en todas las direcciones medidas perpendicularmente desde cualquier punto en su superficie.
a) Dividir la superficie de la pieza a evaluar en un número de planos o partes curveadas (unidades) de forma tal que el área de cada unidad pueda ser calculada geométricamente de valores característicos medidos como la longitud, ancho, altura y radio;
b) Las unidades especificadas en el punto anterior deben ser seleccionadas de forma tal que la distancia entre el área geométrica definida y el material debe ser siempre menor de 3 mm;
c) Determinar la longitud de los valores característicos con una exactitud mayor de 1 mm;
d) Usando la medición de las unidades características, calcular el área geométrica de cada una de las unidades seleccionadas. El área geométrica es la suma de las áreas calculadas de cada una de las unidades, se debe expresar en m2.
2. Determinación del área geométrica de piezas con una superficie geométrica parcial o completamente irregular, piezas que son más delgadas de 4 cm, o piezas generadas por corte de piezas más grandes y que estos lados no van a ser objeto de lixiviación.
Cubrir las partes de la superficie:
Para las piezas en las cuales el área geométrica no puede ser claramente determinada, usando una película resistente al agua.
Aquellas que hallan sido producidas como superficies cortadas o cizalladas, usando una película resistente al agua.
De una pieza a evaluar delgada (con un espesor menor o igual de 4 cm), usando una película resistente al agua, de forma tal que las unidades no cubiertas del área geométrica nunca tengan una distancia mutua menor o igual a 4 cm, medidos perpendicularmente en cualquier punto sobre la superficie geométrica descrita.
Para cubrir, se puede usar un material pegante bueno y resistente al agua (por ejemplo resina acrílica o parafina), aplicado a las superficies de las piezas de ensayo. Determinar el área geométrica remanente después del endurecimiento de la resina.
Dividir la parte no cubierta de la superficie de la pieza a evaluar en un número de planos o partes curvas (unidades), de forma tal que el área de cada unidad pueda ser calculada geométricamente en valores característicos medidos como la longitud, ancho, altura y radio.
Las unidades especificadas en el punto anterior deben ser seleccionadas de forma tal que el área geométrica definida coincida con el área relevante de la pieza a ensayar, donde la distancia actual entre el material y el área definida de la unidad, en el caso de irregularidades en la superficie, debe ser siempre menor de 3 mm.
Determinar la longitud de los valores característicos con una exactitud al menos de 1 mm.
Usando estas, calcular el área geométrica de cada una de las unidades seleccionadas. El área geométrica es la suma de las áreas calculadas de cada una de las unidades; se debe expresar en m2.
3. Piezas a evaluar muy irregulares con ningún lado regular identificable.
a) Cubrir cada superficie de la pieza a evaluar tan ajustadamente como sea posible con una pieza de papel. Use para esto un tipo de papel que no tenga propiedades absorbentes;
b) Mantener el papel alrededor de los bordes de cada una de las superficies de la pieza a evaluar y cortar el papel junto a los pliegues tan exacto como sea posible. También remover cualquier pieza del papel que pueda sobresalir más allá de la superficie;
c) Determinar el peso total de la pieza de papel obtenida del punto anterior;
d) Determinar el peso de la hoja de papel de área conocida y propiedades similares al papel usado en el primer paso:
e) Determinar el área superficial de la pieza a evaluar de la relación de pesos del papel del punto c) y del punto d);
e) Repetir los pasos a) al e) si el ensayo de difusión se va a realizar sobre cuatro o más piezas agregadas juntas como un bache. Determinar el promedio de las mediciones obtenidas. Esta es el área superficial geométrica determinada de acuerdo al método del papel.
Nota: En la determinación usando el método del papel, se puede usar papel para impresora o para fotocopiadoras (hojas A4). Es importante que el papel no tenga cualquier propiedad absorbente al agua. Si la pieza a evaluar es humedecida puede ser necesario secar el papel antes de pesar en el paso c).
VI.ii. Ensayo. Comportamiento del ensayo de difusión
El ensayo se realiza en ocho etapas a una temperatura que puede variar entre los 18 y 22 ºC.
Enjuagar el tanque antes de iniciar el ensayo con ácido nítrico y luego enjuagar con agua desmineralizada. Después colocar la pieza a evaluar en el tanque. Si se coloca más de una pieza en el tanque, el espacio entre las piezas debe ser como mínimo de 2 cm.
1. Etapa 1.
a) Llenar el tanque con una cantidad de agua V determinada con una exactitud de 1% de forma tal que:
Si ninguna parte de la superficie es cubierta:
![]()
Si partes de la superficie están cubiertas:
![]()
Donde:
V es el volumen de fluido lixiviante en litros,
Vp es el volumen de la pieza a evaluar P en litros,
A es el área geométrica de la pieza a evaluar P en m2,
f es un factor de conversión: 1 l/m2.
La pieza a evaluar debe estar localizada de tal forma que todos sus lados estén en contacto con el agua y las partes no cubiertas de las piezas estén sumergidas como mínimo 2 cm;
b) Sellar el tanque;
c) Después de 6 ± 0,5 h, drenar todo el eluato. Esta es la fracción del período 1. No secar o enjuagar la pieza de ensayo;
f) Filtrar sobre un papel filtro de membrana la cantidad de eluato requerido para análisis.
e) Medir el pH (± 0,05).
Nota: El valor del pH y la conductividad son requeridos para determinar si la matriz se ha disuelto durante el ensayo. El valor del pH da una indicación de la alcalinidad de la pieza que se esta evaluando, y el cambio de pH durante el ensayo de difusión da una indicación de la estabilidad del material que se esta investigando. Grandes variaciones en el pH del eluato apuntan hacia que el material no ha alcanzado aún el equilibrio (p.e. no se ha estabilizado aún).
2. Etapa 2 a la 8
Inmediatamente después del drenaje de la Etapa 1, llenar el tanque de nuevo con agua desmineralizada. Usar la misma cantidad V, determinada con una exactitud de 1%, como se usó en la Etapa 1.
Repetir el procedimiento descrito en la Etapa 1 por siete veces como se muestra en la Tabla A.1 (los tiempos aplican desde el momento de la inmersión).
Tabla A.1
Tiempos en los cuales el agua debe ser reemplazada
| Período (n) | Tiempo (días) % |
| 1 | 0,25 ± 10 |
| 2 | 1 ± 10 |
| 3 | 2,25 ± 10 |
| 4 | 4 ± 10 |
| 5 | 9 ± 10 |
| 6 | 16 ± 10 |
| 7 | 36 ± 10 |
| 8 | 64 ± 10 |
Determinar el tiempo de reabastecimiento (el tiempo en el cual el tanque ha sido vaciado) de cada período n, hasta 15 minutos de exactitud.
Al completar el ensayo, pesar el material sólido que puede haberse perdido de la pieza durante el ensayo y que permanezca en el tanque. Este material sólido primero debe ser secado.
Si durante el reabastecimiento se encuentra que relativamente grandes cantidades del material se han perdido de la pieza de ensayo, es recomendado no esperar hasta el final del ensayo; remover el material sólido durante uno o más de los reabastecimientos, secarlo y pesarlo.
Calcular las pérdidas de peso mv (g/m2) del material que se ha perdido de la pieza durante el ensayo (g/A (m2) donde A es el área (no cubierta) de las piezas) en dos fases:
1. Pérdidas de peso mva (g/m2) en la etapa 1 a la 2 del ensayo.
2. Pérdidas de peso mvb (g/m2) en las etapas 2 a la 8 del ensayo.
Nota: Estos dos parámetros dan una visión de las características del material. Grandes pérdidas de peso mva comparadas con mvb indican que la pérdida es una consecuencia de la manera en la cual la pieza ha sido hecha o preparada (por ejemplo, perdidas de un curado inadecuado de la pieza al inicio del ensayo, o pérdidas como resultado de la manera de corta la pieza). Grandes pérdidas de peso mvb comparadas con mva indican que a largo plazo la integridad del material (p.e. pérdidas actuales del material indican moderados enlaces en un material compuesto o pérdidas de efectividad del agente aglomerante bajo la influencia del agua).
XVI. RESULTADOS
A través de este método se presenta la forma de obtener un lixiviado o eluato para residuos monolíticos, se debe medir el pH a dicho lixiviado como se mencionó en el método de medición de pH electrométricamente.
XVII. INFORMACION ESTADISTICA
La información de validación que presenta el método se basa en determinaciones diferentes al pH, ya que como se mencionó el método está basado para la verificación del cumplimiento de los criterios de aceptación de residuos para los diferentes tipos de rellenos de la Unión Europea y no para la determinación de la característica de corrosividad para residuos peligrosos, por tal razón se considera que no son de relevancia los resultados en este caso que se está implementando este método solo para la medición de pH.
XVIII. REFERENCIAS
EA NEN 7375:2004 “Leaching Characteristics of moulded or monolithic building and waste materials. Determination of leaching of inorganic components with the diffusion test – The Tank Test”. Agency Environmental (UK) Based on a translation of the Netherlands Normalisation Institute Standard. Versión 1.0 April 2005.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Figura XI.1. Diagrama de Flujo procedimiento lixiviación materiales monolíticos.
FIGURA EQUIPO EXTRACCIÓN-TCLP/SPLP.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Fuente: Figura 1. SW 846 Método 1311.
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
Fuente: Figura 2. SW 846 Método 1311
EQUIPOS CONOCIDOS POR LA EPA, PARA FILTRACIÓN, ZHE-TCLP/SPLP
APARATOS ROTATORIOS DE AGITACIÓN.
| Compañía | Ubicación | Modelo No. |
| Analytical testing and consulting services, inc. | Warrington, PA, EUA | Extractor 4 contenedores (DC20S) Extractor 8 contenedores (DC20) Extractor 12 contenedores (DC20B) Extractor 24 contenedores (DC24C) |
| Associated design and manufacturing compañy | Alexandria, VA, EUA | 2 contenedores (3740-2-BRE) 4 contenedores (3740-4-BRE) 6 contenedores (3740-6-BRE) 8 contenedores (3740-8-BRE) 12 contenedores (3740-12-BRE) 24 contenedores (3740-24-BRE) |
| Environmental machine and design, Inc | Lynchburg, VA, EUA | 8- contenedores (08-00-00) 4-contenedores (04-00-00) |
| IRA machine shop and laboratory | Santurce, PR, EUA | 8- contenedores (011001) |
| Lars lande manufacturing | Whitmore Lake, MI, EUA | 10-contenedores (10VRE) 5-contenedores (5VRE) 6-contenedores (6VRE) |
| Millipore Corp | Bedford, MA, EUA | 4-ZHE o 4-Extractor de botella de 2 litros (YT310RAHW) |
Extractores de cero espacio de cabeza-ZHE
| Compañía | Ubicación | Modelo No. |
| Analytical testing and consulting services, inc. | Warrington, PA, EUA | C102, equipo de presión mecánico |
| Associated design and manufacturing company | Alexandria, VA, EUA | 3745-ZHE, equipo de presión a gas |
| Environmental machine and design, Inc | Lynchburg, VA, EUA | VOLA-TOX1, equipo de presión a gas |
| Lars lande manufacturing | Whitmore Lake, MI, EUA | ZHE-11, equipo de presión a gas |
| Millipore Corp | Bedford, MA, EUA | YT30090HW, equipo de presión a gas |
| Gelman Science | Ann Arbor, MI, EUA | 15400, equipo de presión a gas. |
Equipo de filtración
| Compañía | Ubicación | Modelo No |
| Nucleopore corporation | Pleasanton, CA, EUA | 425910 (142 mm) 410400 (47 mm) |
| Micro Filtration systems | Dublin, CA, EUA | 302400 (142 mm) 311400 (47 mm) |
| Millipore Corp | Bedford, MA, EUA | YT30142HW (142 mm) XX1004700 (47 mm) |
Medios de filtración
| Compañía | Ubicación | Modelo No. |
| Nucleopore corporation | Pleasanton, CA, EUA | 211625 |
| Micro Filtration systems | Dublin, CA, EUA | GF75 |
| Millipore Corp | Bedford, MA, EUA | AP40 |
| Gelman Science | Ann Arbor, MI, EUA | 66256 (90 mm) 66257 (142 mm) |
| Whatman Laboratory Products, Inc | Clifton, NJ, EUA | GFF |
Métodos apropiados para análisis de analitos en extractos
Métodos SW-846 de análisis recomendados para los Extractos TCLP
< GRÁFICA NO INCLUIDA. VER ORIGINAL EN D.O. No.46.703 - JUL.28-2007 EN LA CARPETA “ANEXOS” O EN LA PÁGINA WEB www.imprenta.gov.co>
3010 Digestión acida de muestras acuosas y extractos para metales totales por análisis por absorción atómica llama o espectroscopia ICP.
3510 Extracción líquido por separador por balón.
3520 Extracción líquido líquido continua.
3535 Extracción de fase sólida (SPE).
6010 Espectroscopia de emisión atómica acoplada a plasma inductivo.
7470 (Ag) Mercurio en desechos líquidos (técnica manual de vapor frío).
8081 Pesticidas organoclorados por cromatografía de gases.
8151 Herbicidas clorados por cromatografía de gases usando metilación o derivatización con pentafluorobenzilación.
8260 Compuestos orgánicos volátiles por cromatografía de gases con detector selectivo de masas (GC/MS).
8270 Compuestos orgánicos semivolátiles por cromatografía de gases con detector selectivo de masas (GC/MS).
8321 Compuestos no volátiles extractables con solvente por cromatografía líquida de alto comportamiento/termoaspersión/espectrometría de masas (HPLC/TS/MS) o detección ultravioleta (UV)
AGUA RECONSTITUIDA.
Ejemplo de un agua de dilución adecuada (de acuerdo a ISO 6341).
Todos los reactivos químicos deben ser grado analítico.
El agua debe ser agua destilada de buena calidad, o desionizada con una conductividad menor de 5 µS cm-1.
El equipo para la destilación del agua no debe contener ninguna parte hecha de cobre.
Soluciones stock.
- Cloruro de calcio dihidratado (CaCl2.2H2O): Disolver 11,76 g en agua y llevar a 1 litro.
- Sulfato de magnesio heptahidratado (MgSO4.7H2O): Disolver 4,93 g en agua y llevar a 1 litro.
- Bicarbonato de sodio (NaHCO3): Disolver 2,59 g en agua y llevar a 1 litro.
- Cloruro de potasio (KCl): Disolver 0,23 g en agua y llevar a 1 litro.
Agua de dilución reconstituida:
Mezclar 25 ml de cada una de las cuatro soluciones stock y llevar a 1 litro con agua.
Airear hasta que la concentración de oxígeno disuelto sea igual al valor de saturación en aire.
El pH debe ser 7,8 ± 0,2.
Si es necesario ajustar el pH con hidróxido de sodio [NaOH] o ácido clorhídrico [HCl].
El agua de dilución preparada de esta forma es dejada aparte por 12 horas y no necesita aireación adicional.
La suma de iones calcio y magnesio en esta solución es de 2,5 mmol por litro. La relación de iones calcio a magnesio Ca:Mg es de 4:1 y la de iones sodio a potasio Na:K es de 10:1. La alcalinidad total de esta solución es de 0,8 mmol por litro.
Cualquier desviación en la preparación del agua de dilución no debe cambiar la composición o propiedades del agua.
SUSTANCIAS DE REFERENCIA.
Resumen de resultados de un ensayo realizado en 1978.
Precaución: el propósito de este ensayo fue la determinación de EC50 en 24 horas.
Sustancias usadas:
1. Dicromato de potasio.
2. Acido tetrapropilbencenosulfónico.
3. Acido tetrapropilbencenosulfónico, sal de sodio.
4. Acido tricloro -2,4,5-fenoxiacético, sal de potasio.
| Sustancia | No De laboratorios participando | No De resultados de calculo | EC50-24 h promedio mg/l |
| 1 | 46 | 129 | 1,5 |
| 2 | 36 | 108 | 27 |
| 3 | 31 | 84 | 27 |
| 4 | 32 | 72 | 770 |
Ejemplo de un procedimiento para el cultivo de algas
Observaciones generales
El propósito descrito a continuación es obtener un cultivo de algas para ensayo de toxicidad.
Se deben utilizar métodos adecuados para asegurar que los cultivos de algas no estén infectados con bacterias (ISO 4833). Monocultivos estériles pueden ser deseables pero cultivos de unialgas son esenciales.
Todas las operaciones deben ser realizadas en condiciones estériles para evitar la contaminación con bacterias y otras algas. Cultivos contaminados se deben rechazar.
Procedimiento para la obtención de cultivos de algas.
Preparación de la solución de nutrientes (medio):
El medio puede ser preparado diluyendo las soluciones stock concentradas de nutrientes. Para medio sólido se adiciona 0,8% de agar. El medio usado debe ser estéril. La esterilización con autoclave puede producir pérdidas de amoniaco.
Cultivo stock.
Los cultivos stock son pequeños cultivos de algas que son regularmente transferidos a medio fresco para actuar como material de ensayo inicial. Si el cultivo no se ha usado regularmente rayar sobre tubos de agar inclinados. Estos deben ser transferidos a medio fresco al menos una vez cada dos meses.
Los cultivos stock deben crecer en recipientes cónicos conteniendo medio apropiado (aproximadamente 100 ml). Cuando las algas son incubadas a 20 ºC con iluminación continua, se requiere transferencia semanal.
Durante la transferencia, una cantidad de cultivo viejo es transferido con una pipeta estéril a un recipiente con medio fresco, de forma tal que para especies de rápido crecimiento la concentración inicial es alrededor de 100 veces menor que en el medio de cultivo viejo.
La tasa de crecimiento de una especie puede ser determinada de la curva de crecimiento. Si esta es conocida, es posible estimar la densidad en la cual el cultivo debe ser transferido al nuevo medio. Esto se debe realizar antes que el cultivo alcance la fase de decaimiento.
Precultivo.
El precultivo tiene como objetivo dar una cantidad de algas adecuadas para la inoculación de los cultivos de ensayo. El precultivo es incubado bajo las condiciones del ensayo y usado cuando se está en fase de crecimiento exponencial, normalmente después de un período de incubación de alrededor de tres días. Se deben descartar las células anormales o deformadas presentes en el cultivo.
RESULTADO DE ENSAYOS INTERLABORATORIOS CON SUSTANCIA DE REFERENCIA.
La ISO 8692-Calidad de agua –Ensayo de inhibición del crecimiento de algas en agua fresca con Scenedesmus subspicatus y Selenastrum capricornutum reporta los siguientes resultados para un estudio interlaboratorios entre 16 laboratorios, evaluando dicromato de potasio.
| Media (mg/l) | Rango (mg/l) | |
| ErC50 (0-72h) | 0,84 | 0,60 a 1,03 |
| EbC50 (0-72h) | 0,53 | 0,20 a 0,75 |
ARTÍCULO 2o. <Resolución derogada por el artículo 35 de la Resolución 63 de 2024> Ordénese la remisión de copia de la presente resolución al Ministerio de Ambiente, Vivienda y Desarrollo Territorial, para su conocimiento, a la Oficina de Informática del Ideam, para su divulgación en la página web del Instituto y al Grupo de Documentación, Archivo y Correspondencia del Ideam, con el fin de que repose en la biblioteca como documento de consulta.
ARTÍCULO 3o. <Resolución derogada por el artículo 35 de la Resolución 63 de 2024> Ordénese la publicación del presente acto administrativo en el Diario Oficial.
ARTÍCULO 4o. La presente resolución rige a partir de la fecha de su publicación.
Publíquese y cúmplase.
Dada en Bogotá, D. C., a 30 de marzo de 2007.
El Director General,
CARLOS COSTA POSADA.
***
1. En caso de que la sustancia problema sea un monolito sólido, el procedimiento de determinación de pH a seguir es el correspondiente al del Anexo I.
2. Electrodos combinados incorporando tanto funciones de medición y referencia son convenientes y son disponibles con materiales sólidos o de llenado tipo gel que requieren mantenimiento mínimo.
3. Para placas con orificios de 1.0 a 8.0 mm de diámetro, deben utilizarse tuercas con orificios de 10.0 mm de diámetro. En caso de placas con diámetros mayores a 8.0 mm el diámetro de la tuerca debe ser de 20.0 mm.
4. Niveles de presión que pueden alcanzarse con determinadas sustancias en los embalajes comerciales normales.
5.En el caso de los equipos automáticos, el procedimiento descrito es realizado por el equipo y el manejo del mismo debe realizarse acorde con las instrucciones del fabricante.
6.Igualmente por lo menos una (1) vez al año debe realizarse la verificación del mecanismo de medición de la temperatura mediante la determinación del flash point de un material de referencia certificado.
7.Si la muestra presenta una viscosidad alta a esta temperatura, debe realizarse un calentamiento previo en el recipiente para muestra antes de su introducción en la cápsula de prueba.
8.En el caso de compuestos halogenados como el cloruro de metileno o el tricloroetileno, no se presenta un flash point distinguible por estos efectos, sino que ocurre una extensión de la llama de la fuente de ignición y un cambio en su color de azul a amarillo naranja.
9. En caso de sustancias a las que se deba aplicar el procedimiento B, realizar los ajustes necesarios al diagrama presentado en las etapas de agitación y calentamiento de la muestra.
10. En el caso de muestras que correspondan a polvos metálicos o aleaciones de metales, se omite esta etapa de la prueba.
11. La solución puede estar formada por agua y agentes humectantes y el contenido de sustancia activa no debe exceder el 1% en la solución.
12. Para la adición de la solución, puede abrirse un pequeño agujero de hasta 3 mm de diámetro y 5 mm de profundidad en la parte superior de la muestra.
13. Para la prueba con este tipo de sustancias debe emplearse atmósfera de nitrógeno en cada una de las etapas comprendidas en el método.
14. Sustancias que tienen la capacidad de activar o aumentar la intensidad de la combustión.
15. Algunos desechos, como los desechos aceitosos y algunos desechos de pinturas, contienen material que parece ser líquido y que después de filtrar por vacío o presión este líquido no filtra. En este caso el material dentro del equipo de filtración es definido como la fase sólida. No se debe reemplazar el filtro original bajo ninguna circunstancia por un filtro nuevo. Usar solo un filtro.
16. Filtros enjuagados con ácido pueden ser usados para todas las extracciones de compuestos no-volátiles aun cuando los metales no sean de interés.
17. Si el desecho (> 1% del peso de la muestra original) se ha adherido al contenedor usado para transferir la muestra al aparato de filtración, se debe determinar el peso de este residuo y restarlo del peso de la muestra para determinar el peso de la muestra que será filtrada.
18. La aplicación instantánea de alta presión puede degradar el filtro de fibra de vidrio y puede causar obstrucción prematura.
19. Algunos desechos, como los desechos aceitosos y algunos desechos de pinturas, contienen material que parece ser líquido y que después de filtrar por vacío o presión este líquido no filtra. En este caso el material dentro del equipo de filtración es definido como la fase sólida. No se debe reemplazar el filtro original bajo ninguna circunstancia por un filtro nuevo. Usar solo un filtro.
20. Durante la agitación, se puede acumular presión dentro de las botellas de extracción con algunos desechos (p.ej. carbonatos de calcio, cal, pueden generar gases tales como dióxido de carbono). Para liberar la presión excesiva, las botellas del extractor pueden ser abiertas periódicamente (p.ej. después de 15 minutos, 30 minutos, y 1 hora) y venteadas dentro de una cabina de extracción.
21. No se recomienda tamizar los desechos debido a la posibilidad de que los compuestos volátiles puedan perderse. Se recomienda como una alternativa aceptable el uso de una regla apropiadamente graduada. Requerimientos de área superficial aplican para desechos filamentosos (p.e. papel, ropa) y materiales de desecho similares. No se recomiendan las mediciones de área superficial actualmente.
22. La aplicación instantánea de alta presión puede degradar el filtro fibra de vidrio y puede causar obstrucción prematura.
23. Algunos desechos, como los desechos aceitosos y algunos desechos de pinturas, contienen material que parece ser líquido y que después de filtrar por presión este líquido no filtra. En este caso el material dentro del equipo de filtración es definido como la fase sólida. No se debe reemplazar el filtro original bajo ninguna circunstancia por un filtro nuevo. Usar solo un filtro.
24. Las soluciones no son buffer y puede que no se obtenga el pH exacto.
25. Se deben verificar impurezas de estos fluidos frecuentemente. El pH debe ser chequeado. Si no cumplen con las especificaciones de pH y presenta muchas impurezas el fluido debe ser descartado y preparar uno fresco.
26. Algunos desechos, como los desechos aceitosos y algunos desechos de pinturas, contienen material que parece ser líquido y que después de filtrar por vacío o presión este líquido no filtra. En este caso el material dentro del equipo de filtración es definido como la fase sólida. No se debe reemplazar el filtro original bajo ninguna circunstancia por un filtro nuevo. Usar solo un filtro.
27. Filtros enjuagados con ácido pueden ser usados para todas las extracciones de compuestos novolátiles aun cuando los metales no sean de interés.
28. Si el desecho (> 1% del peso de la muestra original) se ha adherido al contenedor usado para transferir la muestra al aparato de filtración, se debe determinar el peso de este residuo y restarlo del peso de la muestra para determinar el peso de la muestra que será filtrada.
29. La aplicación instantánea de alta presión puede degradar el filtro de fibra de vidrio y puede causar obstrucción prematura.
30. Para algunos desechos, como los desechos aceitosos y algunos desechos de pinturas, contienen material que parece ser líquido y que después de filtrar por vacío o presión este líquido no filtra. En este caso el material dentro del equipo de filtración es definido como la fase sólida. No se debe reemplazar el filtro original bajo ninguna circunstancia por un filtro nuevo. Usar solo un filtro.
31. Durante la agitación, se puede acumular presión dentro de las botellas de extracción con algunos desechos (p.e. carbonatos de calcio, cal, pueden generar gases tales como dióxido de carbono). Para liberar la presión excesiva, las botellas del extractor pueden ser abiertas periódicamente (p.e. después de 15 minutos, 30 minutos, y 1 hora) y venteadas dentro de una cabina de extracción.
32. No se recomienda tamizar los desechos debido a la posibilidad de que los compuestos volátiles puedan perderse. Se recomienda como una alternativa aceptable el uso de una regla apropiadamente graduada. Requerimientos de área superficial aplican para desechos filamentosos (p.e. papel, ropa) y materiales de desecho similares. No se recomiendan las mediciones de área superficial actualmente.
33 La aplicación instantánea de alta presión puede degradar el filtro fibra de vidrio y puede causar obstrucción prematura.
34. Para algunos desechos, como los desechos aceitosos y algunos desechos de pinturas, contienen material que parece ser líquido y que después de filtrar por presión este líquido no filtra. En este caso el material dentro del equipo de filtración es definido como la fase sólida. No se debe reemplazar el filtro original bajo ninguna circunstancia por un filtro nuevo. Usar solo un filtro.
35. Densidad celular: número de células por mililitro.
36. ATCC = Colección de cultivos tipo americano –American Type Culture Collection (USA)
37. CCAP = Centro de cultivos de algas y protozoos–Culture centre of algae and protozoa(UK).
38. SAG = Colección de cultivos de algas –Collection of algal culture (Gottingen, F.R.G).
39. Referencia 4 del método, S.Galassi and M vighi –Chemosphere, 1981, vol 10, 1123-1126.



